
Abstract
High rates of new cervical cancer cases and deaths occur in low- and middle-income countries yearly, and one reason was found related to limitation of regular cervical cancer screening in local and low-resource settings. HPV has over 150 types, yet certain 14–20 high-risk and 13–14 low-risk types are common, and, thus, most conventional HPV nucleic acid assays, for examples, Cobas 4800 HPV test (Roche Diagnostics, New Jersey, USA) and REBA HPV-ID (Molecules and Diagnostics, Wonju, Republic of Korea) were developed to cover these types. We thereby utilized bioinformatics combined with recent isothermal amplification technique at 35–42 °C to firstly describe multiplex recombinase polymerase amplification assay that is specific to these common 20 high-risk and 14 low-risk types, and also L1 and E6/E7 genes that target different stages of cervical cancer development. Multiplex primer concentrations and reaction incubation conditions were optimized to allow simultaneous two gene detections at limit of detection of 1000 copies (equivalent to 2.01 fg) for L1 and 100 copies (0.0125 fg) for E6/E7, respectively. The assay was validated against urogenital and other pathogens, normal flora, and human control. In 130 real clinical sample tests, the assay demonstrated 100% specificity, 78% diagnostic accuracy, and 75% sensitivity compared with REBA HPV-ID test, and is much more rapid (15–40 min), less expensive (~ 3–4 USD/reaction) and does not require instrumentation (35–42 °C reaction condition so hand holding or tropical temperature is possible). Hence, the developed novel assay provides alternative screening tool for potential local screening. Furthermore, as this assay uses safe chemical reagents, it is safe for users.
Similar content being viewed by others


Introduction
Cervical cancer is a major public health problem in developed and developing countries. According to the World Health Organization (WHO), an estimated 604,000 new cases and 342,000 deaths of cervical cancer occurred worldwide in 2020, and 90% of these cases were in low- and middle-income countries1,2. The reasons were found primarily related to a smaller rate of access to cervical cancer vaccination as a preventive venue and to regular cervical cancer screening programs (and, thus, screening-positive women could be treated adequately before cancer development) compared with high-income countries2. In Thailand and other Southeast Asia countries, cervical cancer represents the most common cancer among women. The Institute of Oncology/International Agency for Research on Cancer reported that ~ 9158 women were diagnosed with cervical cancer and 4705 died every year, and estimated approximately 3.4% of the Thai women population carried human papillomavirus (HPV) high-risk types 16 or 183. In addition, the cost of radiation therapy for cervical cancer and treatment of side effects, such as gastrointestinal or genitourinary side effects, is as high as ~ 63,103 USD per patient. The 2018 economic burden for merely radiation therapy in Thailand was ~ 131 million USD, and for the diagnosis of these advanced cervical cancer patients was ~ 129 million USD4. Subsequently, an effective, simple, easy-access, and low-price screening method for early prevention is important to minimize the number of cases and the economic burden of advanced cervical cancer patients.
More than 95% of cervical cancer are caused by HPV. HPV is also associated with more than 60–90% of anal cancer, vaginal cancer, oropharyngeal cancer, and penile cancer5,6,7. HPV has over 150 types and is classified into high-risk (HR) or low-risk (LR) for cervical cancer development. Common HPV HR types include 16, 18, 31, 33, 34, 35, 39, 45, 51, 52, 58, 59, 68, and 82, given types 16 and 18 generally account for 70% of cervical cancer. Common HPV LR types include 6, 11, 32, 40, 42, 43, 44, 54, 61, 70, 72, 81, 84, and 872,5,8,9.
Traditionally, a cytological examination by Papanicolaou test (Pap smear) is simple and established. However, the procedure is inconvenient, cannot be performed in local resource- and clinician-limited settings, and its sensitivity (i.e., the limit of detection) and specificity to detect precancerous or cancerous changes in cells are relatively low compared with nucleic acid amplification test10,11. For instance, Spence et al.12 reported a false negative rate of Pap smear to be 35.5% on average with the sensitivity ranging from 30 to 87%11. Furthermore, the Pap smear may require another HPV-specific nucleic acid test, such as Cobas 4800 HPV test (Roche Diagnostics, New Jersey, USA) and REBA HPV-ID (Molecules and Diagnostics, Wonju, Republic of Korea), as accompaniment and identification of HR types. Therefore, the HPV-specific tests were suggested as an alternative screening tool and some even reported that these tests could allow screening intervals to be extended to ≥ 5 years11,13,14,15. The Cobas 4800 HPV test (price ~ 8.5 USD, time ~ 5 h per reaction) utilizes amplification of target DNA by polymerase chain reaction (PCR) and nucleic acid hybridization for detection of HPV HR types 16, 18, and a pool of 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 6815. REBA HPV-ID (price ~ 25.2 USD, time ~ 6 h per reaction) is a PCR-based reverse blot hybridization assay for 19 HPV HR probes (types 16, 18, 26, 31, 33, 34, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 69, and 73) and 13 LR probes (types 6, 11, 32, 40, 42, 43, 44, 54, 70, 72, 81, 84, and 87) for HR and LR genotyping16. Although the Cobas and REBA assays are available commercially and used widely in in vitro clinical laboratories, they require an expensive thermal cycler for PCR amplification of HPV DNA and a special instrument for product-probe detection15,16.
Recently, an isothermal amplification assay like recombinase polymerase amplification is emerged to solve this problem. The recombinase polymerase amplification utilizes 3 enzymes, a recombinase, a single-stranded DNA-binding protein, and strand-displacing polymerase, to allow isothermal amplification of specific primer-binding target(s) at 37–42 °C. This allows the reaction to be simply performed by a heat block or water bath, or even holding a tube in hands, supporting low cost, rapid and point-of-care molecular tests17. For HPV detection, Ma et al.18 developed RPA for HPV HR types 16 and 18, and Gong et al.19 developed RPA for 13 HPV HR types based on the L1 gene. We, thereby, further developed a multiplex recombinase polymerase amplification (mRPA) assay with attempt to cover broad HPV HR and LR types, as well as normal and precancerous stages of HPV developmental cycle in humans, as potential local use in a single tube for broad and precancerous screenings of cervical cancer. We determined the limit of detection, validated proper specificity on positive controls comprising some HR and LR types and negative controls comprising urogenital bacteria, other viruses, human and mouse, and evaluated the assay efficiency (e.g., sensitivity, specificity, and accuracy) on real 130 clinical cervical swab samples compared with the Cobas and REBA results. In HPV developmental cycle, a late gene L1 encodes for a major viral capsid protein, while early genes E6 and E7 encode for oncoproteins and their detection suggests an increased risk of cancer development (e.g., the higher E6/E7 than L1 mRNA expression was found in cancer patients). E6 degrades a human growth suppressor protein p53 and E7 degrades a human retinoblastoma tumor suppressor protein pRb, and, thus, the E6 and E7 induce cancer development by evasion of human growth suppressors, resisting cell death, sustaining proliferative signaling, enabling replicative immortality, angiogenesis induction, and activation of invasion and metastasis20. Consequently, this study designed multiplex degenerate primers based on HPV L1 and E6/E7 genes, covering up to 20 HR and 14 LR types, and the E6/E7 amplification product could suggest an increased risk of cancer.
Materials and methods
mRPA primers and design
All GenBank available HPV type L1 (and E6/E7) sequences were aligned using ClustalW (www.megasoftware.net/), and RPA degenerate primers were designed using Primer-BLAST21 or PrimedRPA22 and manual design (Table 1). Of each gene, a cocktail of more than one degenerate primer sequences were used to allow primer coverage of up to 20 HPV HR types (16, 18, 26, 31, 33, 34, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 69, 73, and 82) and 14 LR types (6, 11, 32, 40, 42, 43, 44, 54, 61, 70, 72, 81, 84 and 87). Each primer specificity was verified by BLASTN. The primers were synthesized by Macrogen Inc., Korea.
Reference and clinical strains
Microorganisms used in this study included 12 negative samples comprising bacteria that are sexually transmitted disease pathogens (Chlamydia trachomatis, Neisseria gonorrhoeae, and Staphylococcus saprophyticus), urogenital normal flora (Staphylococcus aureus and Escherichia coli), skin normal flora (Staphylococcus epidermidis), viruses, and mammals (Table 2). These references were provided by the Buddhachinaraj Phitsanulok Hospital; Department of Medical Sciences, Ministry of Public Health; Department of Microbiology, Faculty of Science, Chulalongkorn University; and Faculty of Medicine, Chulalongkorn University23,24,25,26.
For HPV positive controls and clinical samples, the respective genomic DNA were provided by the Buddhachinaraj Phitsanulok Hospital, Phitsanulok; and the Faculty of Medicine, Chulalongkorn University, Bangkok23,27. These sample collections and the protocols were approved by the Institutional Review Board of Buddhachinaraj Phitsanulok Hospital (101/54); and the Institutional Review Board of the Faculty of Medicine, Chulalongkorn University (COA No.278/2019, IRB No. 042/60), and the Institutional Biosafety Committee (IBC) of the Faculty of Medicine, Chulalongkorn University (MDCU-IBC016/2019), respectively. In brief, cervical swab samples were collected by genecologists from patients who visited the Buddhachinaraj Phitsanulok Hospital during 2013–2014 and the Department of Gynecological Out Patient, King Chulalongkorn Memorial Hospital, Bangkok, during 2019–2020. The samples were lysed in Pathogen Lysis Tubes L (Qiagen, Germany) using Tissue Lyser LT (Qiagen), and total nucleic acids were extracted automatically using the Cobas 4800 system (Roche Diagnostics). Then, these genomic DNA were HPV genotyped following the Cobas 4800 HPV test (Roche Diagnostics) and the REBA HPV-ID test (Molecules and Diagnostics); and were also the respective genomic DNA for our mRPA assay. All subjects provided written informed consent, and all data were fully anonymized before assessment. The study was performed in compliance with the Declaration of Helsinki.
Optimization of mRPA reaction condition
The mRPA reaction (25 μL) comprised 0.12–0.48 µM each of primers (unless specified) in Table 1, the TwistAmp® Basic reaction powder (TwistDx, Maidenhead, United Kingdom), 14.75 μL dehydration buffer (TwistDx), ~ 25 ng DNA (unless specified), and 1.25 μL of 280 mM magnesium acetate (pipetted inside the lid and spun down in the final mix step). The reaction was incubated at 39 °C (unless specified) for 40 min (unless specified). The optimization of the mRPA reaction condition included the determination of each minimal primer concentration that yielded both L1 and E6/E7 gene amplification products, and the shortest incubation time with the highest limit of detection (LOD) and correct specificity. The reaction was terminated by 80 °C heating for 2 min. The result was analyzed via 3% agarose gel electrophoresis: the amplicon sizes for L1 and E6/7 RPA were ~ 250 bp and ~ 170 bp, respectively.
Evaluation of RPA primers by PCR and preparation of DNA templates
Single-gene PCR and L1-E6/E7 multiplex PCR (mPCR) were employed as the parallel nucleotide amplification experiments for our mRPA primers and LOD. The PCR reaction (25 µL) comprised 12.5 µL EmeraldAmp® GT PCR Master Mix (TakaRa Bio, Shiga, Japan), 0.12–0.48 µM each primer, and ~ 25 ng DNA (unless specified). The PCR conditions were 95 °C 5 min followed by 35 cycles of 95 °C 15 s, 56 °C 30 s, and 72 °C 30 s, with a final extension at 72 °C 5 min. The proper size amplicon product was analyzed by 3% agarose gel electrophoresis. The amplicon sizes for L1 and E6/7 PCR were 188–194 bp and 117–121 bp, respectively. Quantitation of the amplicon was additionally performed using Qubit™ dsDNA HS and BR Assay Kits (Thermo Fisher Scientific, Massachusetts, USA). This kit quantifies the amount of amplicons (double-stranded DNA, dsDNA) at high sensitivity.
For preparation of HPV L1 (and E6/E7) gene as representative HPV HR and LR DNA templates, the PCR primers (Supplemental Table 1) flanking the RPA amplifying region templates were used. The PCR conditions were similar to the aforementioned, except for the annealing temperature and the product size as listed in Supplemental Table 1. The PCR products were agarose purified and measured quantity using Qubit™ dsDNA HS Assay Kit (Invitrogen). The copy number (copy/µL) was calculated using the following equation: (DNA concentration (ng/µL) × 6.0221 × 1023) ÷ (PCR product length (bp) × 6.6 × 1011).
Comparison of HPV clinical sample diagnosis between our developed methods with conventional Cobas and REBA tests
To determine the performances of the conventional (Cobas and REBA) and our developed methods, statistically required sample numbers (N) (calculator.net/sample-size-calculator.html) were calculated according to the equation: N = (p (1-p) z2)/e2, given p at an average incidence of 75%, confidence level of 95%, and e of 7.5% for margin of error. This yielded an N of 129. Therefore, we tested 130 random HPV-positive and negative samples by our developed mRPA methods compared with the conventional Cobas and REBA results. The assay efficacy was calculated based on the following equations: sensitivity = true positive ÷ (true positive + false negative), specificity = true negative ÷ (true negative + false positive), false positive = 1 − specificity, false negative = 1 − sensitivity, and accuracy = (true positive + true negative) ÷ total samples.
Results and discussion
Specificity of mRPA
As sequences of HPV L1 and E6/E7 are highly variable among types, a primer multiplex system containing 8 degenerate primers was required to cover broad HPV L1 and E6/E7 gene types, of 20 HR (16, 18, 26, 31, 33, 34, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 69, 73, and 82) and 14 LR (6, 11, 32, 40, 42, 43, 44, 54, 61, 70, 72, 81, 84 and 87) types. We performed bioinformatic analyses by BLASTN on each HPV primer sequence specificity against NCBI GenBank non-redundant database (any other viruses, bacteria, mammals and plants), and the primers were specific to HPV (Table 1). Our primers, for example, RPA_L1_F showed no overlap of > 60% and no sequence identity of > 60% to any other viruses, bacteria, mammals and plants. The primers were then PCR checked for the specificity (Fig. 1A). We confirmed the specificity by individual gene RPA, and by mRPA, and the reactions were negative for all tested negative samples including sexually transmitted disease pathogens, urogenital normal flora, skin normal flora, viruses, and mammals (Fig. 1B). Noted that the mPCR and mRPA of the positive HPV HR samples showed only the L1 but not the E6/E7 gene band (only patients no. 11 and 12 showed E6/E6 RPA bands). We compared the %GC among primers and found that the RPA_E6/E7_F contains the relatively high %GC that might affect its higher melting temperature than the other primers and thus the harder to anneal to the template, causing the lower effective in E6/E7 than L1 amplification in mixed templates. Furthermore, in aspect of cancer progression when HPV genome integration occurs in > 80% of HPV-positive cervical cancers, the models of HPV DNA integration that promote oncogenesis known as E6/E7 super-enhancer disrupt the downstream E2 suppressor gene, where located the L2 and L1 genes, in orderly, to give a selective advantage. Breakpoints were often found within the early E1–E5 and late L1–L2 genes, and this inactivation of the E2 repressor upon HPV DNA integration is consistent with the enhanced E6/E7 expression28,29. Hence, the inclusion of E6/E7 in this study benefited the diagnosis in this HPV DNA integration subjects, and the mPCR and mRPA of the positive HPV HR samples that mostly no E6/E7 gene band appeared might reflect the non-precancerous state of the sample.

Specificity of our mRPA multiplex primers for L1 and E6/E7 genes by PCR (A) and RPA (B). Specificity assay included positive (HR types 16, 18, 45, 51, 52, 56, 58, 59, 62, 66; and LR type 6, 42, 54, 70) and negative (C. trachomatis, N. gonorrhoeae, S. saprophyticus, S. aureus, S. epidermidis, E. coli, hepatitis B, SARS-CoV-2, influenza A and B, Homo sapiens, Mus musculus, and sterile water) controls. Grouping of cropped gels were delineated with white spaces, and original uncropped images were in S1 File.
The RPA bands on the electrophoretic image were sometimes slightly skewed up ~ 100 bp might be because the unique auto-strand displacing mRPA amplicon conformations (with recombinase complex and single-stranded DNA binding protein (SSB) in RPA) making the amplicons heavier and we performed a simple one-step clean-up the reaction for low-cost and rapid potential local use, in replace of an amplicon purification step using commercial kits. The shifts of RPA amplicon bands had been reported elsewhere30,31. Noted that in our mPCR and mRPA displayed the < 100 bp bands (< ~ 200 bp for mRPA), which were primer dimers and non-specific amplification, and were found in some positive and negative clinical specificity samples, for examples, influenza B, C. trachomatis, N. gonorrhoaea, S. saprophyticus, S. aureaus, S. epidermidis, H. sapiens and M. musculus (Fig. 1).
Optimal conditions and sensitivity (limit of detection) of mRPA
We first attempted to use forward and reverse primers each at 0.48 µM concentration, but did not work for our mPCR and mRPA. Then, because we had two genes and one-to-more than one forward and reverse primers, we divided in approximate equal and started with 0.1, 0.2 0.3 and 0.4 µM each primer such that the overall concentration of each forward multiplex and each reverse multiplex primers were not much exceeding ~ 0.48 µM concentrations (the recommended concentration for TwistAmp™ Basic Kit). We found the minimal concentration of multiplex primers for L1 (0.1–0.2 µM each) and E6/E7 (0.2 µM each) RPA to be different (Table 3A, Supplemental Fig. 1). In continuing, we tested the L1 and E6/E7 mRPA using a different combination of both gene multiplex primer concentrations to determine the minimal concentration where the mRPA of both genes worked meanwhile the good range of limit of detection. We followed the multiplex primer adjusting methods by Sint et al.32 to obtain equal amplification efficiency. In brief, each gene primer was individually checked for a single amplification fragment, given that synthetic PCR amplicons of HR-HPV types 18 and 33 and LR-HPV type 11 also supported the validation of the effectiveness of the primers (Fig. 2, Supplemental Table 1). Then, the multiplex primers were tested from a gradient of low to higher concentrations with single and mixed templates to determine the optimal concentrations, and some primer concentrations were finally adjusted stepwise to obtain equal amplification efficiency by decreasing those that resulted in relatively strong signal and increasing the ones producing too weak bands in steps of ~ 0.1 µM. We found 0.12–0.24 µM each for the L1 primers and 0.24–0.48 µM each for the E6/E7 primers (Table 3A). For the optimal incubation time, although the amplicons were observed faintly since 15 min, the 40 min incubation allowed clear distinctiveness of both amplicons (Table 3B, Fig. 2A). For an incubation temperature, the RPA enzyme and reaction were suggested at 39 °C by TwistDx, and the optimal incubation temperatures of 35 °C, 37 °C and 42 °C were additionally performed to determine if incubation temperature adjustment was needed. We found that the optimal incubation temperature for mRPA could be 37 or 39 °C due to their relatively strongest bands (Fig. 2B). The limit of detection at the optimal conditions of mRPA was found at 1,000 copies (equivalent to 2.01 fg) for L1 and 100 copies (0.0125 fg) for E6/E7 (Fig. 2C: a faint band of E6/E7 at ~ 170 bp). For clear and confirmatory LOD determination of E6/E7, the mRPA reaction was also tested with a single E6/E7 template and the faint band remained observed at 100 copies (Supplemental Fig. 2). These limits of detection were comparable to some conventional assays, i.e. 1000 copies LOD by Human Papilloma Virus (HPV) DNA Diagnostic Kit (3DMed Diagnostics, Shanghai, China) and 10,000 copies by Primary Screening HPV Test Kit (Jiangsu Mole Bioscience, Jiangsu, China). The obstacles of the lower sensitivity and specificity were often reported for multiplex primers yet to replace with the powerful and cost-effective simple single-tube HPV HR and LR mRPA that is still very critical for the screening purpose in many clinical and epidemiology settings33,34.

Agarose electrophoretic images of L1 and E6/E7 mRPA amplicons at optimal multiplex primer concentrations and incubation time (40 min) (A), incubation temperatures (B), and tenfold serial dilution determination of L1 and E6/E7 limits of detection (C), using mixed templates. In (C), grouping of cropped gels were delineated with white space, and original uncropped images were in S1 File.
To confirm the color depth of the band, we quantified the amount of amplicons at high sensitivity with double-stranded DNA fluorescent dye using Qubit™ dsDNA HS and BR Assay Kits (Thermo Fisher Scientific). Individual L1 (and E6/E7) primers could successfully amplified the HPV templates (Fig. 3A,C). Consistent with the semi-quantitative agarose electrophoretic analyses (Table 3), the fluorescent quantitation of the optimal mRPA primer concentrations demonstrated the relatively greatest fluorescent units (Fig. 3B,D).

Fluorescent quantitation to validate optimal range of primer concentrations in 1-gene PCR reactions (A and C, L1 and E6/E7) and multiplex 2-gene PCR reactions (B and D). In (A and C), individual primers were added at specified 0.1–0.4 µM on X-axis and synthetic L1 (or E6/E7) amplicons at > 108 copies were used as template. For (B and D), primers were added following primer concentration selections from Table 3 and synthetic L1 (or E6/E7) amplicons at 101, 102, 103 and 104 copies were used as template.
Comparison of HPV clinical sample diagnosis of our developed methods with conventional Cobas and REBA tests
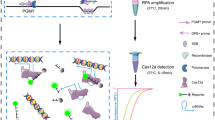
The schematic illustration of RPA and multiplex 2-gene amplification concepts as local HR- and LR-HPV screening tool was summarized in Fig. 4. For potential local use of our single-tube mRPA as broad HPV test (20 HR and 14 LR types) in 40 min, we evaluated the feasibility and compared the assay performance on a statistical number of real clinical DNA samples (N = 130 samples), with Cobas and REBA results. Our developed mRPA assay demonstrated 72–100% specificity, 71–75% sensitivity and 72–78% accuracy, with < 1 h assay time (20–40 min mRPA and 20 min agarose gel electrophoresis) and 2.3–6.7 folds lower price per reaction (Table 4, S2 File). The approximately 75% assay accuracy and 100–10,000 limit of detection are in the reported ranges of other HPV assays and could still serve useful as HPV screening, especially in resource-constrained settings as the reaction requires no specific equipment35,36. Meanwhile, the Cobas and REBA results exhibited 18.46% diagnostic disagreement between each other. Furthermore, the mRPA result analysis by agarose gel electrophoresis may be replaced by using fluorescent DNA dye, such as SYBR Green I (Invitrogen, New York, USA), or DNA probe paper strip for an even more rapid total assay time. We are continuing to improve the mRPA limit of detection by reducing the number of primers to focus on the HPV HR types and the gene expression detection of E6/E7 as reverse transcription-mRPA37.

Schematic illustration of RPA and multiplex 2-gene amplification concepts for potential local applications.
Conclusions
Our developed HPV mRPA assay for broad-type HPV detection (Thai patent pending entitled multiplex recombinase polymerase amplification assay for detection of high risk type and low risk type HPV, with ease, rapidity, low cost, high sensitivity and specificity, in a single tube) is simple, rapid, inexpensive, specific, sensitive, and does not require any complicated instrumentation (incubation may be accomplished by hand holding or appropriate tropical temperature may be possible) (Fig. 4). Evaluation of the assay over positive–negative controls showed proper detections, and on 130 clinical samples compared with the conventional commercial Cobas and REBA assays, both showing 18.46% result disagreement, showed greater than 70% assay effectiveness (72–78% accuracy). The detection limit is as low as 1000 copies (2.01 fg) of the L1 HR and LR types and 100 copies (0.0125 fg) of the E6/E7, and the developed assay could detect at least 20 HPV HR and 14 LR types. Further, the reaction incubation might be performed via hand holding and the alternative fluorescent DNA dye may be used to replace the agarose gel electrophoresis. This developed assay is, thereby, a promising tool for potential screening in local and low-resource settings, to allow disease detection before the cancerous state of the disease.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Stelzle, D. et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob. Health 9, e161–e169. https://doi.org/10.1016/S2214-109X(20)30459-9 (2020).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
ICO/IARC (Institute of Oncology/ International Agency for Research on Cancer) Information Centre on HPV and Cancer. HPV Information Centre. Thailand human papillomavirus and related cancers, fact sheet 2021. https://hpvcentre.net/statistics/reports/THA_FS.pdf.
Katanyoo, K., Riewpaiboon, A., Chaikledkaew, U. & Thavorncharoensap, M. The cost of locally advanced cervical cancer in Thailand: An empirical study for economic analysis. Asian Pac. J. Cancer Prev. 22, 3171–3179 (2021).
Muñoz, N. et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348, 518–527 (2003).
Smith, J. S. et al. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 121, 621–632 (2007).
Combes, J. D. & Franceschi, S. Role of human papillomavirus in non-oropharyngeal head and neck cancers. Oral Oncol. 50, 370–379 (2014).
Burd, E. M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 16, 1–17. https://doi.org/10.1128/cmr.16.1.1-17.2003 (2003).
Vo, D. T. & Story, M. D. Facile and direct detection of human papillomavirus (HPV) DNA in cells using loop-mediated isothermal amplification (LAMP). Mol. Cell Probes 59, 101760. https://doi.org/10.1016/j.mcp.2021.101760 (2021).
Mayrand, M. H. et al. Human papillomavirus DNA versus Papanicolaou screening tests for cervical cancer. N. Engl. J. Med. 357, 1579–1588 (2007).
Boone, J. D., Erickson, B. K. & Huh, W. K. New insights into cervical cancer screening. J. Gynecol. Oncol. 23, 282–287 (2012).
Spence, A. R., Goggin, P. & Franco, E. L. Process of care failures in invasive cervical cancer: Systematic review and meta-analysis. Prev. Med. 45, 93–106 (2007).
Ronco, G. et al. Efficacy of HPV-based screening for prevention of invasive cervical cancer: Follow-up of four European randomised controlled trials. Lancet 383, 524–532 (2014).
Vink, M. A., Bogaards, J. A., Meijer, C. J. & Berkhof, J. Primary human papillomavirus DNA screening for cervical cancer prevention: Can the screening interval be safely extended?. Int. J. Cancer 137, 420–427 (2015).
Liu, S. S. et al. Clinical performance of the Roche Cobas 4800 HPV test for primary cervical cancer screening in a Chinese population. PLoS ONE 17, e0272721. https://doi.org/10.1371/journal.pone.0272721 (2022).
Kim, S. et al. REBA HPV-ID for efficient genotyping of human papillomavirus in clinical samples from Korean patients. J. Med. Virol. 84, 1248–1253 (2012).
Amer, H. M. et al. A new approach for diagnosis of bovine coronavirus using a reverse transcription recombinase polymerase amplification assay. J. Virol. Methods 193, 337–340 (2013).
Ma, B. et al. Isothermal method of a recombinase polymerase amplification assay for the detection of most common high-risk human papillomavirus type 16 and type 18 DNA. Clin. Lab. 63, 27–38 (2017).
Gong, J. et al. A simple and rapid diagnostic method for 13 types of high-risk human papillomavirus (HR-HPV) detection using CRISPR-Cas12a technology. Sci. Rep. 11, 12800. https://doi.org/10.1038/s41598-021-92329-2 (2021).
Pal, A. & Kundu, R. Human papillomavirus E6 and E7: the cervical cancer hallmarks and targets for therapy. Front. Microbiol. 10, 3116. https://doi.org/10.3389/fmicb.2019.03116 (2020).
Ye, J. et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134. https://doi.org/10.1186/1471-2105-13-134 (2012).
Higgins, M. et al. PrimedRPA: Primer design for recombinase polymerase amplification assays. Bioinformatics 35, 682–684 (2019).
Somboonna, N. et al. Rapid and sensitive detection of Chlamydia trachomatis sexually transmitted infections in resource-constrained settings in Thailand at the point-of-care. PLoS Negl. Trop. Dis. 12, e0006900. https://doi.org/10.1371/journal.pntd.0006900 (2018).
Srimongkol, G. et al. Rapid colorimetric loop-mediated isothermal amplification for hypersensitive point-of-care Staphylococcus aureus enterotoxin A gene detection in milk and pork products. Sci. Rep. 10, 7768. https://doi.org/10.1038/s41598-020-64710-0 (2020).
Choopara, I. et al. Fluorometric paper-based loop-mediated isothermal amplification (LAMP) device for quantitative point-of-care detection of methicillin resistant Staphylococcus aureus (MRSA). ACS Sens. 6, 742–751 (2021).
Choopara, I. et al. Specific and sensitive, ready-to-use universal fungi detection by visual color using ITS1 loop-mediated isothermal amplification combined hydroxynaphthol blue. PeerJ 9, e11082. https://doi.org/10.7717/peerj.11082 (2021).
Somboonna, N., Choopara, I., Sukhonpan, K. & Sayasathid, J. Sexually transmitted diseases in symptomatic and asymptomatic Thai females (a study from Bangkok and vicinity). Asian Biomed. 9, 299–311 (2015).
McBride, A. A. & Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 13, e1006211. https://doi.org/10.1371/journal.ppat.1006211 (2017).
Holmes, A. et al. Mechanistic signatures of HPV insertions in cervical carcinomas. npj Genomic Med. 1, 16004. https://doi.org/10.1038/npjgenmed.2016 (2016).
Georgoutsou-Spyridonos, M. et al. Isothermal recombinase polymerase amplification (RPA) of E. coli gDNA in commercially fabricated PCB-based microfluidic platforms. Micromachines 12, 1387. https://doi.org/10.3390/mi12111387 (2021).
Wang, H. et al. Development and evaluation of serotype-specific recombinase polymerase amplification combined with lateral flow dipstick assays for the diagnosis of foot-and-mouth disease virus serotype A, O and Asia1. BMC Vet. Res. 14, 359. https://doi.org/10.1186/s12917-018-1644-4 (2018).
Sint, D., Raso, L. & Traugott, M. Advances in multiplex PCR: Balancing primer efficiencies and improving detection success. Methods Ecol. Evol. 3, 898–905 (2012).
Elnifro, E. M., Ashshi, A. M., Cooper, R. J. & Klapper, P. E. Multiplex PCR: Optimization and application in diagnostic virology. Clin. Microbiol. Rev. 13, 559–570 (2000).
Li, J., Pollak, N. M. & MacDonald, J. Multiplex detection of nucleic acids using recombinase polymerase amplification and a molecular colorimetric 7-segment display. ACS Omiga 4, 11388–11396 (2019).
Hesselink, A. T. et al. Clinical validation of the HPV-risk assay, a novel real-time PCR assay for detection of high-risk human papillomavirus DNA by targeting the E7 region. J. Clin. Microbiol. 52, 890–896 (2014).
Ozaki, S. et al. Analytical performance of newly developed multiplex human papillomavirus genotyping assay using Luminex xMAP™ technology (Mebgen™ HPV Kit). J. Virol. Methods 204, 73–80 (2014).
Weston, G. et al. Use of the Aptima mRNA high-risk human papillomavirus (HR-HPV) assay compared to a DNA HR-HPV assay in the English cervical screening programme: A decision tree model based economic evaluation. BMJ Open 10, e031303. https://doi.org/10.1136/bmjopen-2019-031303 (2020).
Acknowledgements
This research was supported by the Health Systems Research Institute Fund (HSRI), and the Second Century Fund (C2F) and Microbiome Research Unit for Probiotics in Food and Cosmetics, Chulalongkorn University.
Author information
Authors and Affiliations
Contributions
R.W. performed experiments and helped draft manuscript. P.B., A.C., D.W., W.K. and D.D. advised. P.B. and A.C. provided cervical swab DNA samples. N.S., P.A.O., T.P. and A.L. provided negative control samples. N.S. conceived of the study, coordinated and participated in experiments and data analyses, drafted and revised manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wongsamart, R., Bhattarakasol, P., Chaiwongkot, A. et al. Multiplex recombinase polymerase amplification for high-risk and low-risk type HPV detection, as potential local use in single tube. Sci Rep 13, 829 (2023). https://doi.org/10.1038/s41598-023-28038-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-28038-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
