
Abstract
The arachidonic acid (AA) pathway plays a key role in cardiovascular biology, carcinogenesis, and many inflammatory diseases, such as asthma, arthritis, etc. Esterified AA on the inner surface of the cell membrane is hydrolyzed to its free form by phospholipase A2 (PLA2), which is in turn further metabolized by cyclooxygenases (COXs) and lipoxygenases (LOXs) and cytochrome P450 (CYP) enzymes to a spectrum of bioactive mediators that includes prostanoids, leukotrienes (LTs), epoxyeicosatrienoic acids (EETs), dihydroxyeicosatetraenoic acid (diHETEs), eicosatetraenoic acids (ETEs), and lipoxins (LXs). Many of the latter mediators are considered to be novel preventive and therapeutic targets for cardiovascular diseases (CVD), cancers, and inflammatory diseases. This review sets out to summarize the physiological and pathophysiological importance of the AA metabolizing pathways and outline the molecular mechanisms underlying the actions of AA related to its three main metabolic pathways in CVD and cancer progression will provide valuable insight for developing new therapeutic drugs for CVD and anti-cancer agents such as inhibitors of EETs or 2J2. Thus, we herein present a synopsis of AA metabolism in human health, cardiovascular and cancer biology, and the signaling pathways involved in these processes. To explore the role of the AA metabolism and potential therapies, we also introduce the current newly clinical studies targeting AA metabolisms in the different disease conditions.
Similar content being viewed by others

Introduction
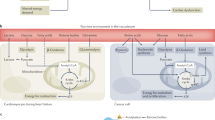
The ω-6 polyunsaturated fatty acid (PUFA), arachidonic acid (AA), and its metabolites have attracted a lot of attention in cardiovascular and cancer biology, particularly in relation to inflammatory processes and disease.1,2,3,4,5,6 The importance of AA in biology lies in the fact that it can be metabolized by three distinct enzyme systems, i.e., cyclooxygenases (COXs, also referred to as PGG/H synthases), lipoxygenases (LOXs), and cytochrome P450 (CYP) enzymes (ω-hydroxylases and epoxygenases) to generate an impressive spectrum of biologically active fatty acid mediators (Fig. 1).

Overview of the arachidonic acid (AA) metabolism pathways. Three major phospholipase enzymes (PLA2, PLC and PLD) are responsible for releasing AA from membrane-bound phospholipids by catalyzing the red arrow indicated covalent bonds, respectively. The PGHSs (COXs) metabolize AA to protanoids, prostacyclin, and thromboxane. The LOXs metabolize AA to leukotrienes and HETEs. The P450 epoxygenases metabolize AA to midchain HETEs and four EET regioisomers. All EETs are then further metabolized to less active dihydroxyeicosatrienoic acids (DHETs) by sEH
The COXs, which generate prostanoids, i.e., prostaglandins (PGs) and thromboxane A2 (TXA2), were the first enzymes reported to metabolize AA. This requires the release of the lipid from the plasma membrane by phospholipases and subsequent metabolism by the COX enzymes to PGG2 and PGH2. The latter are then metabolized to PGs by specific PG synthases. There are two distinct COX isoforms; COX-1, which is constitutively expressed in most cells, is the dominant source of prostanoids that subserve housekeeping functions.7 COX-2 (also known as PTGS2), on the other hand, is induced by inflammatory stimuli, hormones, and growth factors, is generally assumed to be the more important source of prostanoid formation in inflammation and in proliferative diseases, such as cancer.7,8 However, the situation is not black and white as both enzymes contribute to the generation of autoregulatory and homeostatic prostanoids, and both can contribute to prostanoid released during inflammation. Indeed, aspirin and non-steroidal anti-inflammatory drugs (NSAIDs), including inhibitors of COX-2 are effective in the treatment of pain and inflammation.9,10 However, the inhibition PGI2 production by the endothelium may contribute to the cardiovascular side effects of COX2 inhibitors.11 It is thought that inhibition of blood clotting by aspirin can reduce the risk of ischaemic events such as heart attacks and stroke, and prostacyclin analogues are used for the treatment of pulmonary hypertension.9,12,13
The LOX pathway was the second eicosanoid and inflammatory pathway to be therapeutically targeted. The enzymes generate leukotrienes (LTs) which were first described in 1979 by Bengt I. Samuelsson who was awarded the Nobel Prize in Physiology or Medicine in 1982.14 Arachidonate 5-LOX (or ALOX5) and LT receptor antagonists have been developed for the treatment of asthma and seasonal allergies.15,16 These two eicosanoid pathways (COX and LOX) are becoming increasingly important therapeutic targets as novel receptors and metabolites are identified and their roles in many diseases are better defined.
The third AA metabolizing pathway is the cytochrome P450 (CYP) pathway that was first described in 1980. The CYP family of enzymes contains numerous subclasses,17 but for the metabolism of AA ω-hydroxylase and epoxygenase activity are the most important. However, numerous CYP enzymes have mixed hyprolase and epoxygenase functions and are able to generate a mixed spectrum of products. The ω-hydroxylase activity of CYP enzymes converts AA to hydroxyeicosatetraenoic acids (HETEs). 20-HETEs is the best-studied metabolite in this context and has been shown to possess pro-inflammatory effects in addition to contributing to vascular function.18 The epoxygenase activity of CYP enzymes, such as the CYP2J and 2C families, generates AA epoxides or epoxyeicosatrienoic acids (EETs; 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET). Bioactive EETs are produced in the liver with biologically relevant amounts also being detected in the vascularure as well as in cardiomyocytes. The EETs are mainly metabolized by soluble epoxide hydrolase (sEH) to the corresponding diols or dihydroxyeicosatrienoic acids (DHET).19,20 AA diols were initially thought to be less active than the epoxides, but it is now clear that the epoxide and diols may even exert antagonistic actions in some conditions. As the EETs are reported to elicit vasodilatation, this pathway and its metabolites are currently being targeted for the treatment of cardiovascular diseases (CVDs) including hypertension, heart failure (HF), and stroke.21,22 In addition, CYP-derived EETs also regulate some cellular processes of carcinogenesis and progression, including cell proliferation, survival, angiogenesis, invasion, and metastasis. CYP-derived EETs can also promote progenitor cell differentiation, proliferation, and migration, in addition to influencing capillary formation inflammation and apoptosis in endothelial cells. In this way CYP-derived AA metabolites can contribute to tumor growth, progression, and metastasis.23
In this Review, we focus on recent insights into the roles of AA metabolism from molecular mechanisms to clinical studies, particularly in CVD, cancer biology and inflammatory diseases.
Overview of AA metabolism
The COX pathway
As stated above, the term COX refers to enzymes also known as prostaglandin G/H synthases (PGHS), which metabolize AA to PGH2 and PHG2. These PGs are substrates for a series of downstream enzymes that generate specific PGs i.e. PGE2, PGI2, PGD2, PGF2, and TXA2.24,25,26 The major difference between the 2 COX enzymes is that while COX-1 is more or less ubiquitously and constitutively expressed, COX-2 is an inducible enzyme,26,27,28 albeit with some important exceptions.29,30 There are preferences in the coupling between COX and downstream synthases as COX-1 couples preferentially, but not exclusively, with thromboxane synthase, PGF synthase, and the cytosolic (c) PGE synthase (PGES) isozymes. COX-2, on the other hands, more frequently feeds PGG2/H2 to the prostaglandin I synthase (PGIS) and the microsomal (m) PGES isozymes, both of which are often coinduced with COX-2 by cytokines and tumor promoters.31,32,33,34 The profile of prostanoid production is determined by the differential expression of these enzymes within cells present at sites of inflammation. For example, mast cells predominantly generate PGD2, whereas macrophages produce PGE2 and TXA2.35 In addition, alterations in the profile of prostanoid synthesis can occur on cellular activation. An additional COX enzyme, i.e., COX-3, a splice variant of COX-136 that also produced PGH2 has been identified and its expression is reportedly higher in microvessels of the brain and heart than in larger conduit arteries.37,38
PGs exert their effects by activating membrane-localized G protein-coupled receptors and the prostanoid receptor subfamily is composed of 8 members; the PGD receptor (DP1), the PGF receptor (FP); the PGI receptor (IP), the thromboxane receptor (TP), and 4 subtypes of E prostanoid receptor (EP1-4).39 Alternative splicing complicates the situation further and is responsible for two additional isoforms of the human TP (TPα, TPβ) and FP (FPA, FPB) receptors as well as eight variants of EP3 which differ only in their C-terminal tails.40 In addition, there is a distinct G protein-coupled receptor, i.e., chemoattractant receptor-homologous molecule (CRTH2 or DP2) that is expressed on T helper 2 cells that belongs to the family of chemokine receptors but can be activated by PGD2.40,41 Prostanoid receptors couple to a range of intracellular signaling pathways that mediate the effects of receptor activation on cell function. For example, the EP2, EP4, IP, and DP1 receptors activate adenylyl cyclase via Gs, to increase intracellular cAMP whereas EP1 and FP activation couples to phosphatidylinositol metabolism via Gq, leading to the formation of inositol trisphosphate with mobilization of intracellular free calcium.42,43
The LOX pathway
The LOX enzymes insert molecular oxygen in AA and depending on its position, 4 hydroperoxyeicosatetraenoic acids (HPETEs; 5-, 8-, 12-, and 15-HPETE) are formed by the corresponding LOX enzymes, i.e., 5-LOX, 8-LOX, 12-LOX, and 15-LOX. The HPETEs are then reduced into monohydroxy eicosatetraenoic acids (HETEs) by peroxidases, or converted to biologically active compounds such as LTs, lipoxins (LXs), and hepoxilins.
Perhaps the best-studied LOX enzyme is 5-LOX, which inserts oxygen into AA at the C-5 position to generate 5-HPETE and subsequently LTA4, the precursor of the LTs (LTB4, LTC4, LTD4 and LTE4).44,45,46 Although 5-LOX was originally purified as a cytosolic protein, it was later shown to translocate to the nuclear envelope after phosphorylation.47,48 It is now accepted that the nuclear membrane is the major site for the production of LTs. 5-HPETE is further hydrolyzed by LTA4 hydrolase to generate LTB4.48,49 For its catalytic activity 5-LOX requires a 5-LOX-activating protein (FLAP),50,51 a membrane-spanning protein with three transmembrane domains belonging to the “membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG)” family that includes LTC4 synthase and microsomal PGE2 synthase.15,48,52 The precise role of FLAP in 5-LOX reactions remains to be fully elucidated but it is thought that FLAP presents AA to 5-LOX and/or functions as a scaffold for 5-LOX.53 A large body of work now documents the role of 5-LOX-generated products in the pathogenesis and progression of CVD,54 particularly atherosclerosis, MI, stroke, aortic aneurysms, and intimal hyperplasia. 5-LOX-derived mediators in particular focus are oxo-ETEs generated from HETEs by the microsomal dehydrogenase in polymorphonuclear leukocytes (PMNLs), which now seems to be a strong eosinophil chemoattractant.55 Also, LTs are now recognized as a crucial component of vascular inflammation.56 These are generated by is a bi-functional enzyme, i.e., the LTA4 hydrolase—a cytosolic protein that has both LTA4 hydrolase and zinc-dependent peptidase activities. Although the biological role of the LTA4 hydrolase as a peptidase is unknown, it limits pulmonary inflammation by degrading the chemotactic peptide PGP (proline-glycine-proline).57 Thus, in inflammation the LTA4 hydrolase generates a chemotactic lipid mediator, i.e., LTB4, at the same time as degrading a chemotactic peptide, i.e., PGP. Two major pathways of LTB4 inactivation are known, and responsible enzymes have been identified. Granulocytes and hepatocytes inactivate LTB4 through the ω-oxidation pathway58 in which C-20 of LTB4 is oxidized by CYP enzymes; CYP4F3 in granulocytes and CYP4F1 and 2 in hepatocytes.59 In other tissues, LTB4 is inactivated by conversion into 12-keto-LTB4 by the LTB4 12-hydroxydehydrogenase,48,60 which is also involved in the inactivation of various eicosanoids including PG48 and LXA4.61 As far as signaling is concerned, LTC4 exerts its actions on smooth muscle contractions through CysLT1 and CysLT2 receptors. LTB4, on the other hand, acts via LTB4R (BLT1) and LTB4R2 (BLT2) receptors.62
In addition to their ability to generate HETEs via a similar process as described above for 5-LOX, 12-LOX and 15-LOX also generate LXs, oxo-ETEs, and dihydroxyeicosatetraenoic acids (diHETEs).63 For example, 12-LOX can convert 5(S)-HETE to 5(S),12(S)-diHETE as well to 14(R),15(S)-diHETE in the, which ultimately contribute to the generation of extra-platelet LTA4.64,65 The formation of LXs involves 5-LOX in neutrophils and 12-LOX in platelets. In neutrophils, 5-LOX generates LTA4, which is then transferred to platelets where 12-LOX subsequently generates either LXA4 or LXB4.66,67 There are two isoforms of 15-LOX in mammalian cells: 15-LOX-1 and 15-LOX-2. 15-LOX-1 is encoded by the arachidonate 15-lipoxygenase (ALOX15) gene, and the functional enzyme metabolizes AA to LXA4, LXB4, and 15-oxo-ETEs. 15-LOX-2, on the other hand, generates 15-oxo-ETE and 8S-HETE.68,69 The efficiency of 15-LOX-1 is 20 times higher than that of 12-LOX,66 thus when 15-diHPETE is provided as substrate, the primary product catalyzed by 12-LOX and 15-LOX-1 is LXB4.
The CYP pathway
CYP genes encode a super-family of mixed-function monooxygenases, which includes more than 6000 individual enzymes (http://drnelson.uthsc.edu/CytochromeP450.html).70 The best-known role of the CYP pathway is the metabolism of lipophilic xenobiotics, including drugs and chemical carcinogens, as well as endogenous compounds such as steroids, fat-soluble vitamins, fatty acids, and biogenic amines. CYP expression and activity are under the control of hormones, growth factors, and transcription factors. Indeed, different CYP subfamilies can display complex tissue- and development-specific expression patterns. Despite this, CYP2C and CYP2J enzymes can be detected in hepatocytes, cardiomyocytes, vascular endothelial cells, smooth muscle cells, and in some epithelial cells, autonomic ganglion cells, hepatocytes, nerve cells, and islet cells.71 To-date perhaps the most impressive links with biological activity are for EETs in liver, kidney, heart, and endothelial cells.71 Importantly, many genetic and environmental factors alter CYP expression resulting in significant changes in the production or removal of bioactive products.
As far as the cardiovascular system is concerned CYP enzymes are important as they generate by ω-hydroxylated HETEs (6-, 17-, 18-, 19-, and 20-HETE). Perhaps the best studied to these is 20-HETE, which has been linked with vasoconstriction and the regulation of myogenic tone.18 The AA epoxides or EETs, i.e., 5,6-, 8,9-, 11,12- and 14,15-EET, have vasodilatory, cardioprotective, and anti-inflammatory activities and can modulate vascular smooth muscle migration, an important event in vascular remodeling and atherosclerosis. Each of the 4 EET regioisomers has stereoisomers, e.g., 11,12-EET exists as 11(R),12(S)-EET and 11(S),12(R)-EET, and the different stereoisomers can elicit distinct actions.72 The intracellular levels of the EETs are tightly regulated by the activity of the sEH, which generates the equivalent DHETs. The latter has traditionally been considered to be less active than their parent EETs. Relevant human CYP enzymes contributing to the formation of EETs and their distribution are listed in Table 1. Although EETs exhibit some similarities in biological functions, there are differences in their actions to some extent. For example, EETs were found to be a slightly stronger pro-angiogenic factor than other in vivo and in vitro.73,74 CYP-derived EETs are probably best studied with respect to their hyperpolarizing properties as EETs are endothelium-derived hyperpolarizing factors (EDHF) in some organs (particularly in the heart) and thus contribute to the regulation of vascular function.19 It is also now clear that CYP-derived EETs also protect the heart against acute ischemia-reperfusion injury and chronic non-ischemic cardiomyopathy and hypertension.
AA metabolites in CVD
CVD remains a major cause of disability and death in both Western societies and developing nations. As age and co-morbidities, such as obesity and diabetes, become more prevalent in a population both the human health cost and economic burden of these conditions keep increasing. There is compelling evidence of a role for some AA metabolites generated by COX, LOX and CYP enzymes in the development and progression of CVD.75,76,77
Role of COX enzymes and their products in CVD
COXs and CVD
The COX pathway is one of the major treatment targets in atherosclerotic and ischemic heart disease because it affects major pathophysiological features of these diseases, including platelet aggregation, vessel wall tension, and inflammatory processes in atherosclerotic lesions.12 The anti-inflammatory and anti-thrombotic features of aspirin, the only known irreversible inhibitor of COX-1, are primarily related to the suppression of PG and TXA2 synthesis.78,79 Meta-analyses of randomized trials show that the use of aspirin lowers the risk of cardiovascular events by 15% and myocardial infarction (MI) by as much as 30%.80 Beyond effects on platelets, it seems that the COX-1/TXA2 pathway contributes to vascular hypercontractility in atherosclerotic ApoE-deficient mice, targeting this pathway pharmacologically improves endothelial function.81 Aspirin is the only known nonsteroidial anti-inflammatory drug (NSAID) with cardiovascular protective effects but despite its widespread and long-term use, some aspects of aspirin treatment warrant further investigation; such as the interaction between body weight and the effectiveness of aspirin to prevent cardiovascular events.76 COX-2 expression increases with inflammation and although COX-2 inhibitors preserve left ventricular function and dimensions in murine models of MI, these compounds have been found to increase cardiovascular risk in multiple clinical studies. For example, a retrospective cohort study including over 300,000 individuals suggested that the use of two highly selective COX-2 inhibitors; valdecoxib and rofecoxib, was associated with a higher risk of stroke.82 Also, rofecoxib and etoricoxib increased blood pressure, whereas other members of this class of compound, i.e., celecoxib, lumiracoxib, and valdecoxib appeared to have little effect on blood pressure.83 Another retrospective cohort study of over 2000 individuals aged over 65 also indicated a higher combined risk of recurrent congestive HF and mortality in patients prescribed with refecoxib rather than celecoxib.84 The reason for these negative cardiovascular effects seems to be related to inhibition of PGI2 production by the COX-2 expressed by the vascular endothelium exposed to “atheroprotective” laminar flow.85,86 The potent vasoconstrictor 20-HETE is also affected by COX-2 inhibition as it is at least partially inactivated by a COX-2-dependent metabolic step.75,87 Combined therapeutic approaches may get around some of these issues and a new class of drugs that blocks both the COX-1/2 and 5-LOX pathways, may prove to be an effective treatment option for cancer, inflammatory and CVDs, with fewer side effects.88 The compound currently in the most advanced phase of clinical development (phase III) is licofelone, previously known as ML3000.89 Licofelone, characterized as a FLAP inhibitor and also has a weak effect on microsomal prostaglandin E synthase-1 (mPGES-1), developed by Merckle and the partners Alfa Wassermann and Lacer, has reached clinical phase III for the treatment of knee osteoarthritis90 with several studies successfully completed. Compared with other nonsteroidal anti-inflammatory drugs (NSAIDs), licofelone showed improved gastric tolerability and lower incidences of ulcers in healthy volunteers.91
COX products and ischemic cardiomyopathy
A more detailed analysis of the role of prostanoids in the pathogenesis of CVD has been possible with the generation of mice lacking either enzyme involved in prostanoid biosynthesis of prostanoid receptors.12,92,93 Such studies have revealed important and novel roles for prostanoids in the development of acute myocardial infarction (AMI), cardiac hypertrophy, hypertension, atherosclerosis, and vascular remodeling.
PGI2 and TXA2 are the major prostanoids affecting the cardiovascular system and are mainly produced by vascular endothelial cells and platelets.94 Importantly, these compounds are often functional antagonists, i.e., they exert directly opposing effects on a given cell or tissue. This means that the balance in their production is crucial for the maintenance of vascular homeostasis. A shift away from PGI2 towards TXA2 can contribute to the development of various thrombotic diseases.95 Both mediators can also be produced by cardiomyocytes, and their synthesis increased significantly during cardiac ischemia and reperfusion,94,96 suggesting a potential contribution to reperfusion injury. Certainly, PGI2 and its analogues attenuate cardiac reperfusion injury in vivo.97,98 Similarly, TX synthase inhibitors and/or TP antagonists reduce myocardial infarct size in animal studies.99,100
There is evidence for a role of other prostanoids in CVD and PGE2 production also increases during AMI.101,102 What contribution the endogeneously generated PGE2 makes to tissue defence or disease progression has, however, not been determined. More is known about its receptors and even though the expression levels of each EP subtype varied among the species studied, high levels of the EP4 mRNA have been reported in the hearts of several species, including humans.8,103 Using EP4−/− mice it was possible to demonstrate that mice lacking EP4 developed larger infarcts in a model of ischemia and reperfusion. Moreover, isolated perfused hearts (Langendorff preparation) from EP4−/− mice demonstrated more pronounced functional and biochemical derangements in response to ischemia and reperfusion than hearts from wild-type mice.103 EP4 agonists have also been developed and despite the fact that one such compound elicited only weak effects in cardiomyocytes, it markedly increased cAMP concentrations in noncardiomyocytes.103 A second EP4 agonist, significantly reduced infarct size in wild-type mice when administered 1 h prior to coronary occlusion. These results indicate that PGE2 produced endogenously during ischemia or reperfusion can protect the heart from injury.103 Less is known about EP3 receptors but several studies indicate that EP3 agonists also protect the heart from injury by facilitating the opening of KATP channel, also the cardio-äspecific overexpression of EP3 attenuated myocardial ischemia-reperfusion injury.104,105,106
COXs-derived metabolites and cardiac hypertrophy
The role of prostanoids in cardiac hypertrophy has been examined using animal models of pressure overload- and angiotensin II (Ang II)-infusion.107,108 One example is PGI2 as it (and its analogues) can inhibit the Ang II-induced hypertrophy of cultured cardiomyocytes,107 as well as the proliferation and synthesis of collagen by cultured cardiac fibroblasts.109,110 In a more pathophysiologically relevant situation, the PGI2-IP system attenuated the development of pressure overload-induced cardiac hypertrophy by inhibiting both cardiomyocyte hypertrophy and cardiac fibrosis. Specially, the hypertrophic effect of PGF2α on cultured rat cardiomyocytes was not observed in mice due to defective FP signaling.111 Somewhat intriguingly, it seems that PGE2-EP3 is necessary to maintain the normal growth and development of the heart.112 Indeed, the cardiomyocyte-specific deletion of EP3 induces eccentric cardiac hypertrophy and cardiac fibrosis in 16–18-week-old mice, supposedly by inactivating the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway and affecting matrix metal proteinase 2 (MMP-2) expression. Studies on EP4-mediated responses are hampered by the fact that most EP4−/− neonates become pale and lethargic within 24 h of birth and die within 72 h. This phenomenon has been attributed to a failure of the ductus arteriosus to close, and in situ hybridization study showed that EP4 mRNA is strongly expressed in the ductus, suggesting that the receptor plays a role in the regulation of the patency of this vessel.113 Such results also indicate that the normal function of the EP4 receptor is essential for the rapid adaptation of the circulatory system in neonates.113
COXs-derived metabolites and hypertension
Genetic disruption of the EP1 receptor is reported to blunt the acute pressor response to Ang II as well as to reduce chronic Ang II-driven hypertension.114 Also, oral administration of an EP1 receptor antagonist reduced blood pressure in spontaneously hypertensive rats. EP2−/− mice, on the other hand, develop normally but produce small litters and have slightly elevated baseline systolic blood pressures. These animals lacked the characteristic hypotensive response to the intravenous infusion of PGE2, which was in fact converted to hypertension. Such data demonstrate that the EP2 receptor mediates arterial dilatation, salt-sensitive hypertension, and also plays an essential part in female fertility.115 However, PGI2-IP and TXA2-TP system has been reported to be resistant to renovascular hypertension or Ang II-induced hypertension.108,116 In addition, the endothelial expression of PGD synthases, which is responsible for PGD2 synthesis from PGH2, can be upregulated in response to higher shear stress in the circulation.117 Genetic deletion of lipocalin-type PGD synthases in mice triggers hypertension and thrombogenesis.92
Role of LOX enzymes and their products in CVD
During the early phase of inflammation, AA is predominantly metabolized via 5-LOX which generates pro-inflammatory LTs including LTB4. In the later stages of inflammation moving towards resolution PGs, such as PGE2, increase 15-LOX expression which redirects the flow of substrate away from LTB4 synthesis to 15-LOX and the production of LXA4 production. Notably, in vivo levels of LXA4 are decreased in patients with peripheral and coronary atherosclerosis,118 and the overexpression of 12-LOX and 15-LOX in macrophages of atherosclerotic ApoE-deficient mice increase LXA4 production and hamper atherosclerotic lesion development. This atheroprotective effect of LXA4 has been linked to its anti-inflammatory capacity, as it impairs the production of various pro-inflammatory cytokines, stops neutrophil chemotaxis, and induces pro-resolving macrophages functions.78,119,120 Interestingly, aspirin enhances LXA4 production ensued by alleviation of atherosclerotic lesions in ApoE deficient mice.121 Efferocytosis, especially the clearance of polymorphonuclear cells (PMNs) and foam cells, is of major importance for the resolution of inflammation, and its impairment leads to prolongation and progression of inflammatory situations including atherosclerosis. LXs produced by LOX enzymes contribute to the process of efferocytosis.122 Moreover, the protective role of most widely used statin therapies in CVD seems to be (at least partly) attributable to LXA4. Indeed, atorvastatin123 and simvastatin124 can increase the myocardial content of LXA4 and 15(R)-epi-lipoxin-A4 (15-epi-LXA4), both AA products with strong anti-inflammatory properties.125 Despite this, the atheroprotective functions of 12/15-LOX-derived metabolites remain controversial, as 12/15-LOX-ApoE-double-deficient mice were found to be less prone to atherogenesis than ApoE−/− littermates with fully functional 12/15-LOX enzymes.126
In contrast to the mainly atheroprotective roles attributed to the LXs, LTs promote the progression of hyperlipidemia-dependent vascular disease and are associated with atherogenesis, CVD, MI, and stroke.15,127,128 In addition, LTB4 and CysLTs are likely to contribute to the pathophysiology of atherosclerosis and myocardial dysfunction. Accordingly, enhanced activity of the 5-LOX pathway was detected in atherosclerotic lesions,129 and the quantity of 5-LOX positive cells correlated with atherosclerotic lesion progression and plaque stability.78,129 Fitting with this, blocking LTB4 receptors protects against the development of atherosclerosis in ApoE−/− mice,130 and the endothelial overexpression of endothelial cysteinyl leukotriene 2 receptor (CYSLTR2) increase vascular permeability, myocardial ischemia/reperfusion damage, and cardiomyocyte apoptosis in peri-infarct areas.78,131,132 LTB4 also fosters the recruitment of neutrophils to atherosclerotic plaques and contributes to plaque destabilization.133 In line with the pro-atherogenic effects of LTs, they are implicated in myocardial ischemia and reperfusion injury. Accordingly, CYSLTR2 expression within the heart and vasculature is induced by ischemia/reperfusion injury.134 The interaction of LTs with CYSLTR2 increases vascular permeability and amplifies the extent of the myocardial injury, and high levels of CYSLTR2 expression in the heart and vessels have been linked to a detrimental outcome in murine ischemia/reperfusion models.78,134 In line with this, pharmacological blockade of LTBR4 reduces infarct size in a murine model of myocardial ischemia/reperfusion injury,135 and the CYSLTR antagonist; montelukast, which is mainly used in the treatment of asthma and allergic rhinitis, was recently evaluated for its possible cardio-protective effects. Interestingly, both animal models and clinical trials demonstrated a preventive role of montelukast against the development of atherosclerosis and suggested a cardioprotective function.136,137,138
Roles of CYP enzymes and their products in CVD
CYP-derived EETs and heart functions
It is well established that the epoxides of AA generated by CYP enzymes possess biological activity and affect a wide spectrum of cellular and tissue responses as well as having effects on the cardiovascular system. Perhaps most work on the EETs has been performed on vessels and vascular cells and less is known about the actions of cardiac-specific CYP-derived EETs in heart physiology and pathophysiology (Fig. 2), compared with the cardiac expression of CYP subfamilies identified in mammalian species including CYP1A, CYP1B, CYP2A, CYP2B, CYP2D, CYP2E, CYP2J, CYP2R, CYP2S, CYP2U, CYP4A, CYP4B, CYP4F, and CYP11B.139

The main biological functions of CYP-EETs on the cardiovascular system and the main corresponding cellular signaling pathways. CYP epoxygenase metabolites of AA, EETs, act as lipid mediators eliciting numerous biological responses and impacting both vascular and cardiac function, including anti-apoptosis, anti-inflammation, vasodilation, inducing angiogenesis, anti-hypertension and aginst ischemic cardiomyopathy or non-ischemic cardiomyopathy
CYP-derived EETs and ischemic cardiomyopathy
Ischemic cardiomyopathy is defined as CVD resulting from a period of low oxygen flow to the heart.140 This could be due to a blockage resulting in limited blood flow, and consequently oxygen, to the heart. Reduced oxygen levels lead to a wide range of consequences for heart activity and morphology that are detrimental to proper function and homeostasis.140 Overall, CYP-derived EETs in the heart has been shown to improve the outcomes of ischemia and/or ischemia/reperfusion injuries.141,142 This is relevant inasmuch as the expression of many CYP enzymes is increased by hypoxia,143 while that of the sEH is decreased144—conditions that would favor EET stability and bioavailability.
Myocardial ischemia/reperfusion (IR) injury occurs when the coronary flow is obstructed, resulting in widespread damage and remodeling of the heart. MI is characterized by extensive fibrosis, remodeling, inflammation, and myocardial apoptosis that eventually progresses to HF and mortality. The immune system plays an important role in the physiopathology of MI, the increased number of circulating inflammatory leukocytes can lead to more cells accumulating in the myocardium.145 Upon accumulating in the heart, neutrophils, and monocytes participate actively in the inflammatory cascade. Neutrophils do not persist in the infarcted myocardium for very long; their numbers decrease after 3 days, and they almost entirely disappear after 7 days. However, neutrophils have been shown to improve cardiac healing by promoting macrophage polarization towards a reparative phenotype through the release of neutrophil gelatinase-associated lipocalin.146 Thus, although antibody-mediated depletion of neutrophils does not affect infarct size, it does worsen cardiac function and HF, and it also increases cardiac fibrosis.146 In contrast to neutrophils, monocytes continue to accumulate in the ischaemic heart and differentiate to cardiac macrophages for several days. The bone marrow maintains leukocyte production but also expels HSPCs, which accumulate in extramedullary sites such as the spleen. Therein, these cells begin to produce monocytes and neutrophils via a process known as extramedullary hematopoiesis, which increases the number of immune cells that can be recruited to the heart.147 Over the course of several days, the inflammatory phase gives way to a reparative phase,148 which is dominated by the disappearance of neutrophils and the appearance of Ly6Clow macrophages. The transition from inflammation to repair is driven by decreased production of inflammatory cytokines, growth factors, and chemokines.
The common method of inducing MI in vivo is through left anterior descending artery (LAD) occlusion.149,150 This results in a clear and defined infarct region and mimics much of the injury and functional deficits are seen post-MI in humans. Ex vivo models include isolated Langendorff or working heart models to induce IR injury. In vitro, hypoxia/reoxygenation models are typical, although not exactly equivalent to ischemic injury since lack of blood flow in vivo comes with other consequences.139
Models that increase EET bioavailability in mice include the cardiomyocyte-specific overexpression of the human CYP2J2 in C57/BL6 mice, an intervention that improved left ventricular recovery after ischemia and reperfusion.151 Moreover, EET augmentation (mainly 11,12-EET)151 has shown beneficial effects on the chronic effects of such injury. Specifically, preventing the metabolism of EETs by the sEH improves the murine myocardial ejection fraction following LAD ligation and has also been associated with improved myocardial perfusion.152 Similarly, administering EETs for as long as a week following infarction is associated with a reduction in fibrosis. The potentially protective actions of the EETs involve the inhibition of apoptosis, the promotion of pro-survival signaling as well as preserved mitochondrial structure and function. Recently, the endothelial cell-specific overexpression of CYP2J2 was found to improve cardiac function by promoting angiogenesis via Jagged1/Notch1 signaling in a mouse model of LAD ligation. This fits with earlier in vitro studies showing that 11,12-EET and also other EETs induces more robust tube formation and markedly increased vascular endothelial growth factor (VEGF)-A74 and basic fibroblast growth factor (bFGF) expression in hypoxia and normoxia,142 indicating that CYP2J2 in endothelium also contributed to cardioprotection. Moreover, isolated mouse hearts treated directly with EETs or dual-acting compounds possessing EET mimetic and sEH inhibitory properties had reduced infarct size and preserved left ventricular developed pressure (LVDP) compared to controls.142,153 There is evidence to indicate that the protective effect of CYP-EETs on ischemia-reperfusion injury may be age-dependent as the cardioprotective effect of CYP2J2 overexpression declined significantly in old (11–13 months) mice.154 While the molecular events active by the EETs that underlie such protective mechanisms are unknown, results from rat, mouse, and canine models have provided consistent evidence to suggest that the activation of the KATP channels and phosphatidylinositol-3 kinase (PI3K) signaling are involved in EET-associated cardioprotection.155,156 PI3Ks are members of a family of lipid kinases that phosphorylate the 3′-hydroxyl group of phosphatidylinositol (PIP) and PIP2 at the third position, to form PIP2 and PIP3, which activate downstream kinases such as AKT and glycogen synthase kinase 3 (GSK-3β), which during ischemia-reperfusion injury results in reduced cell death and infarct size.157
CYP-derived EETs in non-ischemic cardiomyopathy
In broad terms, non-ischemic cardiomyopathy is myocardial injury leading to arrhythmia, ventricular dysfunction, and HF that is not directly associated with AMI.158 Causes of NICM are complicated and varied including drug toxicity, genetic predisposition, infection, haemodynamic pathology, and immunologic abnormalities.158 Several models are often employed to induce NICM in in vivo, such as transverse aortic constriction (TAC), a surgical model used to stimulate pressure-induced HF, or infusion of Ang II or isoprenaline to induce cardiac hypertrophy and failure.139,159 EETs have demonstrated significant cardioprotective effects in models of non-ischemic cardiomyopathy unrelated to their use as anti-hypertensive agents.160,161 In fact, CYP-derived EETs and sEH inhibitors may represent a promising therapeutic approach for combating detrimental cardiac remodeling and decline of cardiac function, which is a hallmark of NICM. For example, the cardiomyocyte-specific overexpression of CYP2J2 to increase epoxide levels attenuated Ang II-induced cardiac hypertrophy and remodeling via a mechanism dependent on AMPKα2 and a subsequent increase in atrial natriuretic polypeptide (ANP),161 which acts as a vasodilator as well as an inhibitor of fibrosis and renin/aldosterone secretion.162 Importantly, ANP mRNA levels were found to be upregulated 6–14 fold in the myocardium following the AAV-mediated overexpression of CYP2J2 in spontaneously hypertensive rats, an effect that was associated with increased ANP expression in the myocardium and elevated plasma levels of the peptide.163 The relationships described were causative as 11,12-EET stimulated the γ1 domain of the AMP-activated protein kinase (AMPK) α2β2γ1 to bind directly with the protein kinase domain of AKT1, thus accelerating its translocation to the nucleus resulting in increased expression of ANP and abrogation of cardiac hypertrophy.161 In addition, cardiomyocyte-specific expression of CYP2J2 or treatment with EETs protects against cardiac remodeling.160 In Ang II-infused mice overexpressing CYP2J2 specifically in cardiomyocytes, cardioprotection was linked with the activation of peroxisome proliferator-activated receptor (PPAR)-γ, reduced oxidative stress, a decrease in nuclear factor (NF)-κB p65 nuclear translocation, and inhibition of transforming growth factor (TGF)-β1/Smad pathway.160 Similarly, when ISO or Ang II were used to induce cardiac fibrosis, hypertrophy, and dysfunction, beneficial consequences of CYP2J2 overexpression were linked to attenuated NF-κB activation.164 In in vitro experiments, 11,12-EET attenuated cardiomyocyte hypertrophy and the expression of remodeling-related proteins, i.e., collagen I, TGF-β1, tissue inhibitor of matrix metallopeptidase-1 (TIMP1), by similar oxidative stress, NF-κB, PPAR-γ pathway. In an Ang II-induced model of non-ischemic cardiomyopathy, the inhibitory effects of CYP2J2 on cardiac fibrosis were associated with reduced activation of the G12 family Gα proteins (Gα12/13),165 which play a pivotal role in regulating the phenotype of cardiac fibroblasts.166 The latter studies fit well with numerous in vitro and in vivo reports linking the anti-inflammatory properties of EETs with inhibition of the IκBα kinase (IKK)-NF-κB cascade activated by tumor necrosis factor-α.167,168,169 Additional mechanisms attributed to EETs in models of agonist-induced HF has linked CYP2J2 and EETs with reduced endoplasmic reticulum (ER) stress and apoptosis cumulating in improved systolic and diastolic function.170 While EETs can directly affect cardiomyocytes, it is clear that other cardiac cell types are also targeted by EETs, e.g., 14,15-EET treatment suppressed the cardiac inflammatory response, at least in part by preventing macrophages activation.164 Interesting data investigating the protective response of EETs toward LPS-induced cardiac dysfunction also revealed decreased NF-κB activation and the upregulation on PPARα/γ and hemeoxygenase-1 (HO-1) to promote the pre-resolution macrophage phenotype.171 In an experimental approach to increase the biosynthesis of endogenous EETs, overexpression of CYP2J2 in both cell culture and mouse models, attenuated cardiac dysfunction arising from systemic inflammation caused by TNF-α administration.169
Preventing the metabolism of EETs to DHETs by inhibiting the sEH prevented AngII-induced cardiac hypertrophy, in fact, there is a lot of evidence linking AngII with increased sEH expression.172 In a TAC mouse model, beneficial effects of sEH inhibition were noted in the prevention of ventricular arrhythmias that occur in association with cardiac hypertrophy.173 Similarly, sEH-deficient mice that underwent either TAC- or Ang II-induced hypertrophy demonstrated preserved cardiac function compared to controls. Importantly, the sEH−/− mice displayed a stable sinus rhythm with prolonged cardiac repolarization, indicating a protective effect of gene ablation on cardiac arrhythmias.174 Comparable studies in mice with the cardiomyocyte-specific over-expression of CYP2J2 and subjected to TAC or ISO infusion revealed that enhanced cardiac EET biosynthesis is protective against electrical remodeling, ventricular tachyarrhythmia, and atrial fibrillation associated with cardiac hypertrophy.175 The increased survival rate observed in CYP2J2 transgenic mice is attributed to better cardiac electrical stability as only moderate improvements were observed in pump function or hypertrophy.175 Other studies using sEH inhibitors as an approach to increase the bioavailability of EETs and increase EET-mediated cardioprotective effects have demonstrated similar benefits in models of cardiac hypertrophy and HF.176,177 Animal models investigating EET-mediated cardioprotection in models of NICM are becoming more common. However, as with many of the CYP-derived eicosanoids, clinical data remains scarce, and truly translational studies are required to determine whether the CYP-sEH pathway is a safe and manipulatable target for human therapy.
CYP-derived EETs and diabetic cardiomyopathy (DCM)
Metabolic syndrome and diabetes begin an inflammatory cascade that is crucial to the development of cardiomyopathy. Individuals with either type 1 or type 2 diabetes mellitus (T1DM or T2DM) are at greater risk for cardiovascular complications and resultant mortality in non-diabetic subjects.178,179 While diabetes alone carries a risk for heart disease, T2DM is often coupled with other comorbidities such as obesity and metabolic syndrome that additionally complicate the prevention, treatment, and prognosis of patients that go on to develop DCM.178 DCM describes diabetes-related changes in the heart that are separate from CAD and hypertension associated forms of CVD. In diabetes and DCM, inflammation plays a key role and leads altered endothelial cell function, which in turn promotes vascular remodeling, resulting in atherosclerosis and ischemia. Eventually, inflammation induces cardiomyocyte apoptosis, leading to more profound cardiomyopathic changes. At the cellular level, studies have shown that the myocardium suffers from altered substrate utilization, lipotoxicity, increased oxidative stress, and interstitial fibrosis. Lipotoxicity basically describes the storage of fat in organs other than adipose tissue and plays a key role in these events and is also a contributing factor to the development of insulin resistance. Diabetic hearts have decreased myocardial GLUT4, glycolysis, and glucose oxidation, while there is a coincident increase in pyruvate dehydrogenase kinase, β-oxidation, and myocardial oxygen consumption, all of which reflects an increase in fatty acid metabolism secondary to decreased glucose utilization.180 In db/db and ob/ob mouse models of T2DM, the myocardium undergoes changes in mitochondrial morphology and develops mitochondrial uncoupling, leading to reduced ATP synthesis.
As lipid mediators involved in inflammation, hypertension, and glucose homeostasis, EETs are a viable method to protect against DCM. Also, in this situation, the cardiac-specific overexpression of CYP2J2 has beneficial effects on DCM and insulin resistance in high-fat diet-fed, low-dose streptozotocin-treated mice.181 In particular, the overexpression of CYP2J2 resulted in the maintenance of contractile activity, improved heart-specific glucose uptake, and insulin sensitivity, and attenuated the hypertrophy associated with diabetes. Also in this case, the molecular mechanisms underlying these effects were related to insulin-like growth factor 1 (IGF-1), insulin receptor substrate-1 (IRS-1), PI3K, AKT, AMPK, and PPARγ. CYP2J2 over-expression also attenuated increased PDK4 expression, which has been suggested to contribute to DCM by decreasing the pyruvate dehydrogenase complex.181
Ultimately, these studies suggest EETs retain their cardioprotective effects in DCM and may be a useful therapy for patients diagnosed with co-morbidities of diabetes and CVD. Finally, further research in this area is needed to determine whether EETs can be utilized in humans as a cardioprotective strategy against DCM.
CYP-derived EETs and vascular function
Local vascular tone is determined by a variety of factors such as neurotransmitters released from autonomic nerves, circulating vasoactive compounds, tissue metabolites, and endothelium-derived autacoids. The best-characterized vasodilator autacoids are nitric oxide (NO) and prostacyclin (PGI2), but a substantial component of the vasodilator response observed in response to receptor-dependent agonists or increases in flow is insensitive to inhibitors of NO synthases and COXs. Since the NO/PGI2-independent vasodilatation originally described was co-incident with vascular smooth muscle hyperpolarization, and was abolished by depolarizing concentrations of potassium, it was proposed to be mediated by an “EDHF”.182 Campbell et al.19 first reported that EETs relax precontracted coronary artery smooth muscle cells, induce cell hyperpolarization by increasing the open-state probability of Ca2+-activated K+ channels, and identified EETs as an EDHF. Shortly thereafter, the downregulation of a CYP2C enzyme in porcine coronary arteries was demonstrated to abrogate, NO, and PGI2-independent relaxation and hyperpolarization.183 Subsequent studies have demonstrated that the hyperpolarizing effects also exist in peripheral arteries,184 which indicated that CYP-derived EETs elicit vasodilation and improve vascular function in many stress conditions.
CYP-derived EETs and blood pressure
Hypertension is the most prevalent CVD and afflicts one in every three adults worldwide.185 Several factors contribute to chronic blood pressure elevation, which increases the risk for cardiovascular morbidity and mortality. Contributing factors to hypertension include elevated activity of the renin-angiotensin system, increased sympathetic activity, and inflammation.185 These factors result in excessive vasoconstriction and increased total peripheral resistance or impaired sodium excretion, increased extracellular fluid volume, and increased cardiac output.22 In many types of hypertension, EET levels are reported to decrease, an effect attributed to an increase in vascular sEH expression.177
The contribution of CYP eicosanoids to high blood pressure and the associated risk factors has been evaluated in hypertensive animal models as well as in humans. Overexpression of CYP enzymes attenuates the development of hypertension and improves cardiac function in spontaneously hypertensive rats, partly by EGF receptor (EGFR)-dependent effects on ANP.163 Human studies provide evidence that decreased EET levels result in elevated blood pressure,186 as CYP2C gene variants generate fewer EETs and are positively correlated with an increased risk for essential hypertension.187 Consistent with all these findings, increasing EET levels in animal models of hypertension decreases blood pressure and exerts cardiovascular protective actions.177 It therefore seems safe to say that decreased EET production (especially when associated with increased AngII) appears to be a contributing factor to hypertension.177,188,189
It is not just altered vascular production that contributes to hypertension, as CYP enzymes and the sEH are also expressed in the kidney and affect naturists. There is extensive evidence for an important contribution for EETs in maintaining kidney vascular and epithelial function.18,190,191 For example, EETs act to dilate preglomerular afferent arterioles and inhibit epithelial sodium channels (ENaC).192 A decrease in EET levels leads to excessive afferent arteriolar constriction and enhanced ENaC activity and salt absorption, which increases blood volume and blood pressure.193 Indeed, 11,12-EET can inhibit cortical collecting duct ENaC and increase sodium excretion. Conversely, EETs can lower blood pressure by inhibiting sodium absorption in the proximal tubule and cortical collecting duct.194 Importantly, excessive afferent arteriolar constrictor reactivity in hypertension is eliminated by sEH inhibition to increase kidney EET levels.191 Some models of hypertension can even be linked to changes in specific CYP enzymes asn salt-sensitive hypertension occurs when the kidney and vascular expression of CYP2C23 and CYP2C11 fail to increase in response to a high salt diet.191 In accordance with these findings, the genetic deletion of CYP2C23 (CYP2C44) in mice results in decreased kidney and vascular EET levels and salt-sensitive hypertension.
CYP-derived EETs, atherosclerosis, and coronary artery disease
Polymorphisms in the CYP2J2 gene have been shown to affect CAD risk and incidence in specific populations.195,196 One of the most relevant polymorphisms in terms of frequency and functional importance is located at −50 (G-50T) in the proximal promoter of CYP2J2. Screening of 289 patients with coronary artery disease and 255 control subjects revealed 77 individuals with the G-50T SNP (17.3% of CAD patients, 10.6% of control subjects; P = 0.026). The association of the G-50T polymorphism remained significant after adjustment for age, gender, and conventional cardiovascular risk factors (OR, 2.23; 95% CI, 1.04–4.79). The G-50T mutation resulted in the loss of binding of the Sp1 transcription factor to the CYP2J2 promoter and resulted in a 48.1 ± 2.4% decrease in CYP2J2 promoter activity (P < 0.01). Plasma concentrations of stable EET metabolites were significantly lower in individuals with the G-50T SNP.195 In addition, the presence of the CYP2J2*7 allele in an African-American population was associated with a significantly lower risk of incident CAD, while an increased risk of CAD along with lower plasma EET levels were observed in a Caucasian population195 Interestingly, EPHX2 polymorphisms have been linked to risk for coronary artery calcification and disease in young adults.197
In atherosclerosis-prone apolipoprotein E (ApoE)-deficient mice, recombinant adeno-associated virus (rAAV)-mediated CYP2J2 gene overexpression, which is associated with increased EET levels, prevented the development of high-fat diet-induced atherosclerosis.198 Mouse models of atherosclerosis have been relatively extensively studied and treating ApoE−/− mice with sEH inhibitors prevents atherosclerosis induced by a high cholesterol diet.199 Similarly, studies in sEH−/− mice have demonstrated a contribution for EETs to oppose vascular inflammation, atherosclerosis, and vascular remodeling.177 Moreover, sEH-/- mice and animals with endothelial cell-specific overexpression of CYP2C8 or CYP2J2 demonstrate decreased vascular inflammation and NF-κB activity when exposes to endotoxin.18 EET-positive actions to attenuate atherosclerosis has been associated with decreased adhesion molecules and inflammatory cytokines.18 Thus, EETs and sEH inhibition decrease inflammation and have vascular protective actions that can combat atherosclerosis. The effects extend to abdominal aortic aneurysms.200 In particular, CYP2J2 overexpression could be linked with attenuated matrix metalloproteinase expression and activity, elastin degradation, and AAA formation, which was associated with reduced aortic inflammation and macrophage infiltration. Again, these effects were linked with the activation of PPARγ,200 but the same mice also manifested lower low-density lipoprotein and elevated high-density lipoprotein cholesterol levels, as well as attenuated expression of pro-inflammatory genes and proteins.201 These effects were associated with a reduction of serum lipid, interleukin (IL)-6, murine IL-8-KC, and IL-1α, and downregulation of gene expressions of ICAM-1, VCAM-1, and IL-6 in the arterial wall.200,202,203
Collectively, the beneficial effects of EETs and sEH inhibitors in the preclinical model were vasodilation, anti-hypertension, anti-inflammation, improved endothelial function, and lipid regulation. Moreover, the clinical association of sEH gene polymorphisms towards increased risks of atherosclerotic vascular disease provides a strong rationale to target sEH in the treatment of atherosclerosis.204
CYP-derived EETs and stroke
EETs or sEH inhibition protects either the heart or brain from the damage that occurs following an ischemic event.21,152,156,205 This protective action for EETs appears to be multifactorial and EETs likely inhibit apoptosis in the brain tissue. Brain tissue EET cell signaling antiapoptotic mechanisms involve increased Bcl2, ceramide inhibition, and decreased ROS.156,206 Indeed, we found that CYP2J2 overexpression increased EET productions, increases regional cerebral blood flow (rCBF) and microvascular density, decreased ROS production, decreased brain infarct size and apoptosis after ischemia, the effects of which were associated with increased activation of the PI3K/AKT and apoptosis-related protein in the ischemic brain. Liu et al.207 found that exogenous administration of 14,15-EET or AUDA could suppress astrogliosis and glial scar formation, inhibit microglia activation and inflammatory response, promote angiogenesis, attenuate neuronal apoptosis and infarct volume, and further promote the behavioral function recovery after focal ischemia.
Moreover, sEH was widely expressed in spinal cord tissue, mainly confined to astrocytes, and neurons. Administration of sEH inhibitor AUDA significantly suppressed local inflammatory responses as indicated by the reduced microglia activation and IL-1β expression, as well as the decreased infiltration of neutrophils and T lymphocytes.208 Furthermore, treatment of AUDA improved angiogenesis, inhibited neuron cell apoptosis, alleviated demyelination and formation of the cavity and improved motor recovery.208 In addition, epidemiological data demonstrating genetic polymorphism in the EPHX2 are associated with increased risk for ischemic stroke.197 We firstly found that there was a significant interaction between the EPHX2 G860A polymorphism, smoking and ischemic stroke risk such that nonsmokers carrying the EPHX2 G860A variant allele were at the lowest risk of ischemic stroke.209
These results together suggest that epoxyeicosanoid signaling and she inhibition are promising multi-mechanism therapeutic targets for the treatment of stroke.
CYP-derived EETs and angiogenesis
Angiogenesis is a complex process that involves the proliferation, invasion, and migration of endothelial cells to form tubes or primitive capillaries. Epoxides of AA have a clear link to angiogenesis.74,210,211 Munzenmaier et al.212 firstly found the link of CYP-EETs/sEH axis and angiogenesis, in which EETs promoted proliferation and tube formation in cerebral capillary endothelial cells released by cultured astrocytes. This fit well with observations that the overexpression of CYP2C9 and the corresponding production of EETs promoted the activation of the mitogen-activated protein 1 (MKP-1) mediated dephosphorylation and inactivation of c-Jun N-terminal kinase (JNK), effects ultimately culminating in the expression of cyclin D1 and proliferation in human endothelial cells.213 In addition, 11,12-EET-induced transactivation of the EGF receptor and activation of Akt kinase were inhibited by sphingosine kinase (SK) specific inhibitor.214 Activation of AKT by EETs was also linked to PI3K, inhibition of the forkhead factors FOXO1 and FOXO3a and subsequently a decrease in the expression of the cyclin-dependent kinase inhibitor p27kip1. The transfection of CYP2C9 overexpressing cells with either a dominant-negative AKT or a constitutively active FOXO3a inhibited CYP2C9-induced endothelial cell proliferation.215 In addition to the PI3K/AKT pathway, the inhibition of MAPKs was also found to attenuate EETs-induced endothelial proliferation.74 Work from Capdevila’s team further underscored that activation of p38 MAPK is required for the proliferative responses to 8,9- and 11,12-EET, and activation of PI3K is necessary for the cell proliferation induced by 5,6- and 14,15-EET.216 Moreover, treatment with EETs and the sEH inhibitor trans-4-[4-(3-adamantan-1-ylureido)cyclohexyloxy]benzoic acid50,51 (t-AUCB), respectively, significantly increase VEGF production,217 an effect prevented by CYP inhibitors.218 That is, multiple signaling pathways are involved in pro-proliferation effects of CYP-EETs/sEH system on endothelial cells.
Meanwhile, it is important to note that angiogenesis can be stimulated when EETs are generated by endothelial cells themselves, as well as when they were applied exogenously or generated from astrocytes. This indicates that the actions of the EETs cannot be restricted to an autocrine role but that a sufficient EET concentration must be able to leave the cell of origin to elicit paracrine actions on other cells. The development of novel transgenic animals has helped to confirm the effects of CYP-derived metabolites of AA on angiogenesis and vascular repair, e.g., in an ischemic rat hind limb model in which the overexpression of different CYP enzymes, including CYP2C11 and 2J2, was found to increase muscle capillary density.74 However, it remains unclear whether these pathways are linked to each other or are simply activated in parallel.
Endothelial cell migration is an essential step to form vessel-like structures.219 EETs promote endothelial cell migration by a mechanism thought to involve the endothelial NO synthase, MAPK, and the PI3K activation.74,220 The situation appears to be somewhat different in murine pulmonary endothelial cells in which 5,6- and 8,9-EET (but not 11,12- or 14,15-EET) evoke a MEK/MAPK and PI3K-dependent cell migration.216 Prior to migration out of a preexisting mature vessel, endothelial cells need to degrade the surrounding extracellular matrix and inhibit migration and proliferation of vascular smooth muscle cell,221 thus in turn providing space for the migration of endothelial cells and the diffusion of key growth factors, such as FGF-2, PDGF, and VEGF.222,223 A series of enzymes including collagenases, gelatinases, stromolysins, metalloelastases, and membrane-type matrix metalloproteases (MT-MMP), are responsible for the degradation of the extracellular matrix.222 Both 11,12- and 14,15-EET have been reported to activate one or more metalloproteases220,224 and promote the release of heparin-binding EGF-like growth factor (HB-EGF) from the cell surface.225,226 In addition, the sEH inhibitor (12-(3-adamantan-1-yl-ureido)-dodecanoic acid or AUDA) also reduced the protein expression of MMP-9 in ECs227 and MMP activity was increased in CYP-2C9-overexpressing cells increased and correlated with invasion ability.220
The formation of cord-like structures and primitive tubular structures are more direct evidence for angiogenesis. The overexpression of CYP2C9 in and/or the application of 11,12- or 14,15-EET to monocultures of endothelial cells have been linked to the formation of such structures in vitro on matrigel or in fibrin gels.226,228 The in vivo data also rapidly supported these and EETs-induced angiogenesis in the chick chorioallantoic membrane,226 as well as in EET-impregnated matrigel plugs in adult rats228 and in an ischemic rat hind limb model. In these models above, the overexpression of different CYPs, including CYP 2C11 and 2J2, was found to increase muscle capillary density.74 The potential mechanisms of EET-induced angiogenesis include that inhibition of the forkhead transcription factor to downregulate p27Kip1,215 crosstalk to EGF receptor,226 induction of FGF273 and VEGF,229 often demonstrated via AKT activation,215,226 SRC-activation of STAT3,229 the activation of sphingosine kinase-1,214 and the induction of endothelial nitric oxide synthase.74,219 Moreover, EET-induced angiogenesis also involves crosstalk with other AA metabolizing pathways as 11,12-EET induced the expression of COX-2 in endothelial cells via a PKA-cAMP-dependent pathway230 and COX-2 protein shifted EET metabolism away from DHETs and towards epoxy hydroxyeicosatrienoic acids (EHETs) which have been attributed angiogenic properties.231 Which of these pathways is applicable probably depends on the species, type of endothelium, and EET regioisomers produced by the CYP epoxygenase.232
Other non-negligible events in the process of angiogenesis are an adaptation to hypoxia and the differentiation of endothelial precursor cells. This is particularly relevant in the tumor microenvironment (TME) when the pO2 drops once a tumor grows beyond a size where O2 needs can be met by discussion from outside the tumor. Hypoxia stimulates the expression of a series of CYP enzymes in endothelial cells including CYP2C8 and CYP2C9 to increase EET formation.220,233 Importantly, the same stimulus suppresses the expression of the sEH, at least in the mouse liver and a human hepatoma cell line234 to further increase EET levels. Consistently, hypoxia-induced angiogenesis in vitro was abolished by antisense oligonucleotides directed against CYP2C enzymes as well as by the CYP inhibitor MS-PPOH and the EET antagonist 14,15-epoxyeicosa-5(Z)-enoic acid (EEZE)220,233 and enhanced by the endothelial cell-specific overexpression of CYP2J2 or by sEH inhibitors around the ischaemic area in MI model.142,235 Exogenous EETs may even improve diabetic/non-diabetic wound healing caused by ischemia via modulating inflammation and angiogenesis.224,236 Endothelial precursor cells arising from hematopoietic stem cells in the bone marrow; upon proangiogenic stimuli, they proliferate, migrate, and differentiate into mature endothelium in several diseases such as myocardial ischemia, stroke, and in tumor growth and progression.237 Increasing EETs levels with t-AUCB promoted EPCs activation in the AMI patients via a PPARγ dependent manner.238 In addition, aerobic exercise modulated circulating EPC function via elevating EET concentrations in mice with AMI239 Thus, CYP-derived EETs promote angiogenesis via various mechanisms.
CYP-derived HETEs in CVD
CYP enzyme-dependent ω-hydroxylation of AA is a prototypic metabolic reaction of CYP4 family members that is important for hydroxyeicosatetraenoic acid generation. 20-hydroxyeicosatetraenoic acid (20-HETE) is the main product of the reaction catalyzed by three main CYP4 enzymes, i.e., CYP4A11, CYP4F2, and CYP4F3B. Multiple researches have linked 20-HETE with cardiovascular disorders and renal system. 20-HETE has been suggested to mediate androgen-induced hypertension through increasing the level of Cyp4a12240 and the overexpression of the Cyp4a12-20-HETE synthase in proximal tubular promotes salt-sensitive hypertension in male mice.241 In the kidney, however, 20-HETE exerts anti-hypertensive effects through inhibition of sodium reabsorption in the proximal tubule and thick ascending limb of Henle.242 Furthermore, 20-HETE acts as a vasoconstrictor of vascular smooth muscle cells by promoting calcium entry into cells to enhance phosphorylation of contractile elements.243,244 Several studies have suggested an interplay between 20-HETE and the renin–angiotensin aldosterone system (RAAS) in hypertension. Briefly, angiotensinogen II has been reported to increase renal production of 20-HETE, and 20-HETE can activate the RAAS by inducing angiotensin-converting enzyme.245,246,247 CYP4A was also reportedly upregulated in models of doxorubicin-induced cardiotoxicity with a consequent increase of 20-HETE synthesis.248 Furthermore, Jarrar et al.249 found that heart cyp4a12 was highly upregulated in mice after cardiac toxicity induced by NSAIDs. Thus, targeting of 20-HETE synthesis through manipulation of CYP4 enzymes could be considered in the future development of the drug for CVDs.
EET receptors
A mount of data has contributed to the characterization and understanding the role of CYP-derived metabolites function within CVD. However, the identity of the specific receptor(s) involved in epoxylipid responses remains unclear. Given that high-affinity EET binding sites on the surface of some cells, such as monocytes, vascular smooth muscle cells, and endothelial cells, many investigators have speculated that a specific EET receptor may exist on the membrane of cells.182 For instance, the 11(R),12(S)-EET is a more potent activator of renal artery KCa channels250 than 11(S),12(R)-EET. Also, in endothelial cells 11(R),12(S)-EET could induce the membrane translocation of TRPC channels rapidly while the other EETs (such as 14,15-EET and 11(S),12(R)-EET) were ineffective.182 In addition, many evidences suggest the actions of EETs are in part mediated via G-protein-coupled receptor (GPCR) signaling. For instance, biochemical studies have already indicated the importance of Gs Proteins in 11,12-EET-initiated signaling,251 and in endothelial cells the downregulation of Gs but not Gq/11 was recently shown to abrogate the effects of 11(R),12(S)-EET on TRPC6 channels.252 In addition, in HEK293 cells, G protein-coupled receptor 40 (GPR40) was also reported to be involved in mitogenic responses to EETs.253 GPR40 is an interesting candidate EET receptor, in which the medium and long-chain fatty acids are thought to be putative ligands. However, it remains inconclusive whether EET-induced changes in cAMP signaling as a response to classical GPCR cellular responses.252 In addition, it has been reported EETs can induce vasodilation via antagonizing thromboxane (TP) receptors in the vascular system.
Numerous reports illustrate the effects of PPARα and PPARγ activation with EETs. PPARs are involved in regulating lipid metabolism, inflammation, immune function, cell proliferation, and insulin secretion.139,182 Therefore, it is more than likely that these intracellular lipid mediators interact with intracellular receptor molecules such as the PPAR family of nuclear receptors. The significance of PPAR activation in mediating effects of EETs needs further investigation to draw a clear mechanistic pathway.
AA metabolisms in cancer
Cancer is a major health burden worldwide and represents one of the leading causes of mortality and morbidity, with ~14.1 million new cases and 8.2 million cancer-related deaths annually.254 Despite the advance in various treatment strategies, such as combinations of surgical resection, radiation or chemotherapies and immune therapies, the 5-year survival rate for some cancers is still relatively low. Furthermore, the underlying cause(s) of cancer remain unclear. Thus, there is an unmet need to develop an effective strategy for preventing the development of this devastating disease. While the results of large chemoprevention trials thus far have not been encouraging, a 20-year follow-up study with aspirin, a non-steroidal anti-inflammatory agent that acetylates and inhibits COX-2, showed that the mortality rates from all solid cancers were 20% lower for those receiving aspirin, with adenocarcinoma being the most reduced (34%).255,256 This is strong evidence for the role of anti-inflammatory agents such as COX inhibitors in cancer prevention. Probably the COX metabolites with the highest tumorigenic and metastatic potential is PGE2, as it inhibits cancer cell apoptosis and increases invasiveness as well as promoting angiogenesis257 in tumors. The pathways implicated include mTORC1/VEGR, NF-κB, MAPK/JNK/p38, PI3K/Akt as well as epigenetic modifications.258,259,260 There also seems to be a role for CYP-derived EETs in the development of various cancers.261,262
Roles of COXs and their metabolites in cancer
Chronic inflammation is clearly associated with an increase in the risk of cancer.263 One of the strongest associations between chronic inflammation and cancer is the increased risk in individuals with inflammatory bowel diseases. Inflammation also appears to have an important role in the development of other cancers, for example, prostate, bladder, and pancreatic cancers. Chronic inflammation causes the upregulation of a number of inflammatory cytokines including IL-1β, IFNγ, and TNFα. The NF-κB pathway is activated in many chronic inflammatory states, and evidence directly links the NF-κB pathway to increased tumor formation and inflammation in experimental mouse models of intestinal cancer.264,265,266 Because NF-κB plays a role in COX-2 regulation at the transcriptional level, prostaglandin H synthase or COX-2 expression is increased, and higher levels of inflammatory PGs are formed.267 Diminished expression of 15-prostaglandin dehydrogenase (15-PGDH), a prostaglandin degradation enzyme also contributes to the elevated PG levels in cancer.266,268 The aberrant AA metabolism observed in cancer cells results in a high concentration of PGs, in particular, PGE2.41,269 Because of the high concentrations of PGE2 in tumors, many investigations have focused on the EP receptors.266,270 Indeed, EP2 expression is upregulated compared with normal tissues in colorectal and breast cancers.116,266,271 Moreover, both EP2 and EP4 mRNA was upregulated in human glioblastomaastrocytoma U373 MG cells compared to the primary astrocytes.272 The deletion of the EP2 receptor in APC/Min mice substantially reduced polyp formation,271 while deletion of the EP4 receptor has been shown to decrease the formation of aberrant crypt foci in animals treated with the colon carcinogen azoxymethane.273 At the level of signaling, the EP2/4 receptors are G protein-coupled receptors and PGE2 can thus activate PKA to stimulate several divergent pathways to mediate pro-tumorigenic activities.274 For example, PKA phosphorylates GSK-3, to alter the APC/β-catenin/TCF pathway, which regulates cell proliferation, angiogenesis, and apoptosis.256,274,275 PGE2 also can transactivate the EGF receptor, increase amphiregulin, and enhance the RAS-MAP kinase pathway, and transactivate the PPAR δ pathway.276,277,278,279
Numerous epidemiological, clinical, laboratory, and animal and cell culture studies confirm that the use of COX inhibitors or nonsteroidal NSAIDs is effective at inhibiting the incidence and mortality of colorectal cancer.280,281 In addition to colorectal cancer, NSAIDs have also been associated with a reduced risk of breast, esophageal, stomach, bladder, ovary, and lung cancers.282,283,284 Despite the extensive studies on the effectiveness of NSAIDs as chemopreventative agents, the molecular mechanisms underlying their chemopreventative effects are not well understood. While is was initially presumed that the anti-cancer activity of the NSAIDs could be attributed to the inhibition of COX-1/COX-2, this concept has been challenged by the fact that very high doses of COX inhibitors are frequently required to exhibit tumor inhibitory effects but only low doses are required to prevent PG generation.266,285 Therefore, COX-independent effects may contribute to the chemopreventative activity of NSAIDs.285 There is at least circumstantial evidence for such an effect as NSAIDs inhibit the growth of colon cancer cell lines that do not express COX-1 or COX-2286 and inhibit the growth of mouse embryo fibroblasts lacking both the COX-1 and COX-2 genes.287
Roles of LOXs and their metabolites in cancer
The inhibition of COX activity by NSAIDs makes the substrate, i.e., AA, available for metabolism by other enzymes and may cause a shift in the AA metabolite profile from PGs to LOX-derived hydroxylated lipids. 5-LOX, 12-LOX, 15-LOX-1, and 15-LOX-2 are reported to have some influence on tumor development. For example, there are numerous reports of increased 5-LOX expression in cancer cells, e.g., 5-LOX and 5-LOX activating protein (FLAP) was universally expressed in epithelial cancer cell lines,288 and 5-LOX was elevated in human pancreatic cancer cells289 as well as in malignant tissue from patients with prostate carcinoma. The latter study reported 2.2-fold greater levels of 5-HETE in malignant tumor tissue compared with benign tissue.290 Fitting with this. MK591, a specific 5-LOX inhibitor-induced apoptosis in prostate cancer cells via downregulation of PKCε, a pro-survival serine/threonine kinase.291 Similarly, both 5-LOX mRNA and protein were higher in gastric cancer than non-tumor tissues and 5-LOX inhibition induced apoptosis in the human gastric cancer AGS cell line.292 Added to all this, the combined use of the 5-LOX inhibitor zileuton and the COX-2 inhibitor celecoxib elicited synergistic effects in human oral cancer and colon cancer suggesting that COX-2/5-LOX inhibitor may be a more effective direction of antitumor drug discovery.293,294 Indeed, licofelone, a potent COX-2/5-LOX inhibitor was shown to induce apoptosis in both androgen-dependent and androgen-independent prostate and colon cancer cells.295,296
15-LOX-1 is present in human colorectal cancer cells216 and converts AA to 15-HETE and linoleic acid to 13-hydroxyoctadecadienoic acid (13-HODE). Interestingly, 15-LOX-1 has been associated with anti-tumorigenic activity in human colorectal cells,297 and in human colorectal cancer.298 It is perhaps not surprising therefore that the expression of 15-LOX-1 is lower in human colorectal tumors than in normal tissue, and as a consequence, so are the levels of the major 15-LOX-1 metabolite, 13-HODE.266,299 How 13-HODE its anti-tumor effect is likely related to its ability to downregulate PPARδ,300 and stimulate the phosphorylation of the tumor suppressor gene p53, which results in increased expression of many downstream targets.301 However, while the growth inhibitory effects of 15-LOX-1 were p53 dependent, 15-LOX-1 metabolites failed to induce its phosphorylation and a 15-LOX-1 inhibitor did fail to prevent p53 phosphorylation.301 Such findings may indicate that an additional protein may be involved—the interaction of the 15-LOX-1 protein with the DNA-PK kinase which can phosphorylate p53302 could account for such a phenomenon.
12-LOX is the LOX isoform expressed in epithelial cells and myeloid cells including platelets. Many mutations in this isoform are found in epithelial cancers, suggesting a potential link between 12-LOX and tumorigenesis.303 Thus, the LOX, especially 15-LOX-1, appears also to have a role in the reduction of tumors by COX inhibitors.
Recently, Haribabu et al. showed reduced CD8+ T cell migration and increased tumor growth in BLT1−/− mice injected with B16 melanomas, indicating the important role of BLT1 signaling in immune surveillance and anti-tumor immunity.304,305 In the murine spontaneous colon cancer model (ApcMin mice), the same authors also reported that BLT1−/− ApcMin/+ mice showed increased intestinal tumor development, exacerbation of colon inflammation, and increased mortality.304,306 Furthermore, in acrystalline silica-induced lung cancer model, LTB4 production by inflammatory leukocytes increased macrophage phagocytosis and led to sustained activation of neutrophils via an autocrine loop of LTB4 production. Although LTB4-BLT1 signaling was shown to play a key role in anti-tumor responses, critically, the cell-specific roles of BLT1 in vivo are still unknown, and further studies that employ conditional cell-specific knockout of BLT1 are needed in these cancer models.
In addition, LTC4 and its metabolites LTD4 and LTE4 (together referred to as cysteinyl LTs, CysLTs) are inflammatory mediators derived from AA via the 5-LOX pathway.1 They exert many of their functions through the CysLT1 receptor, which is expressed in pulmonary smooth muscle and interstitial macrophages. CysLTs contribute to cancer progression and several observations support a pro-tumorigenic effect of LTD4 via CysLT1 in colorectal cancer.307 Montelukast is a CysLT1 receptor antagonist already used in asthma treatment.308 Interestingly, asthma patients treated with montelukast have a considerably lower risk to develop cancer.309 In animal studies, montelukast increased survival rates in a spontaneous metastasis model of Lewis lung carcinoma (LLC) and delayed tumor growth.308,310
Roles of CYP dependent monooxygenases and their metabolites/sEH in cancer
Emerging evidence has demonstrated that CYP-derived EETs regulates multiple cellular processes of carcinogenesis and progression, including cell proliferation, survival, angiogenesis, invasion, and metastasis.23,311,312 CYP enzymes, such as CYP2J2 are highly expressed in various human carcinoma cell lines (including LS-174, ScaBER, SiHa, U251, A549, Tca-8113, Ncl-H446, and HepG2) and human tumors (including esophageal adenocarcinoma, pulmonary carcinoma, breast carcinoma, stomach carcinoma, liver carcinoma, and colon adenocarcinoma). In animal models CYP2J2 overexpression promoted cancer growth and metastasis,261 and CYP enzyme-derived EETs enhance tumor cell motility, invasion, adhesion and metastasis.262 These studies were a prelude to a wave of subsequent studies reporting the relationship of the CYP-EET/sEH axis and cancer development.
Levels of CYP-EETs in cancer
The high expression level of CYP enzymes implies that AA epoxide levels should also be increased in tumors. EETs were first detected in colonic adenocarcinoma homogenate back in 1995.313 However, because of the chemical instability the measures of the level were much lower than the actual values. This problem led some researches to use DHET levels as an indirect index of EET content, an assumption that certainly held true for cancer cell lines versus HEK-293 cells.261 Also, DHET levels were significantly higher in urine and plasma from patients with leukemia/lymphoma than from healthy volunteers.314 The elevated DHET could indirectly imply elevated sEH activity or expression and pretreatment with sEH inhibitors has been reported to significantly improved the stability of EETs in various types of biological samples.315,316 Since these early reports the development of methodology, especially ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLCMS/MS) has made EET analyses more specific and sensitive.317,318 Using such techniques, EETs have been detected in tumor tissues or cells in various conditions. Conversely, inhibition of EETs generation is the key link to uncover novel approaches for tumor treatment.319,320
Polymorphisms of CYP epoxygenases in cancer
Genetic polymorphisms of CYP enzymes and the sEH, including single nucleotide polymorphisms, gene duplications, and deletions, resulting in abolished, reduced, altered, or increased expression and activity (Table 2). It is worth emphasizing that changes in AA-derived EETs and anti-tumor drugs due to CYP polymorphisms have been related to cancer susceptibility, tumor characteristics, and treatment response. Therefore, there is no doubt that CYP polymorphisms are closely associated with cancer fate.
CYP2J2: At least 9 variants of CYP2J2 have been identified, i.e., CYP2J2*2 to *10 (http://www. https://www.pharmvar.org). CYP2J2*2, *3, *4, and *6 carry A4274G, C4724T, T5754A, and A12104T mutations, leading to a statistically significant decrease in AA metabolism in vitro. These mutations result in 59%, 41%, 30%, and 5% of the wild-type CYP2J2 activity, respectively.321,322 CYP2J2*7 has a G4T substitution in the regulatory region at position-76 (50) of transcription start and lacks the binding site for Sp1, and consequently lowering CYP2J2 protein and its metabolites in vivo.193,195 CYP2J2*8 was reported in Koreans with frequencies of 0.8% in 2005,323 and has a point mutation in exon 6 (G9344A), resulting in the almost complete loss of enzyme activity.323 CYP2J2*10 carries a C3444A mutation in exon 2 was found in one fetal tissue with unknown ethnicity in 2006 and possibly severely damaged CYP2J2 protein activity.251 In contrast, there is no apparent difference between CYP2J2*5 and *9 which carry the G10244A and P3514L mutations, and wild-type CYP2J2—at least as far as AA metabolism is concerned.322,323 Most research on the relationship between CYP2J2 polymorphism and disease has focused on the cardiovascular system instead of neoplastic disease. For example, two intronic CYP2J2 SNPs (rs10889160 and rs11572325) were associated with an increased risk of MI.324 In addition, the most common variant (CYP2J2*7) with the frequency of 1.1–1.2% in a Russian, 2.6% in Chinese, and 11–17% in Africans increased the risk of hypertension and MI,195,325 is linked with a diminished capacity to synthesize EETs.326 Tumor development and cardiovascular benefits are often contradictory when assessing CYP2J2-EET functions. Thus, we speculate that the loss of function of CYP2J2 polymorphism, which detrimental to cardiovascular health, may decrease the risk of neoplastic disease.
CYP2C8: CYP2C8, which constitutes 7% of total hepatic microsomal,327 is responsible for the oxidative metabolism of at least 5% commonly used clinical drugs, including the anticancer drugs paclitaxel, cyclophosphamide and ifosphamide and Imatinib.328,329,330 Regardless of the frequency, 14 polymorphic variants in CYP2C8, referred to as CYP2C8.2 through CYP2C8.14 (http://www.cypalleles.ki.se/cyp2c8.htm) and an unclassified form named CYP2C8 P404A331 have been reported. Of these, CYP2C8*2 (805A > T, Ile269Phe) and CYP2C8*3 (416G > A/1196A > G, Arg139Lys/ Lys399Arg) are two major variant alleles with 4–18% frequency in Africans and 10–23% frequency in Caucasians, respectively.332 Both variants demonstrate decreased enzymatic activity for paclitaxel 6a-hydroxylation, leading to a corresponding increase in drug exposure in paclitaxel-treated patients.330,333 Patients carrying CYP2C8*3 are more likely to achieve complete clinical response to neoadjuvant paclitaxel treatment but have the risk of severe peripheral neurotoxicity.334 The N-demethylation of imatinib, the key drug for patients with chronic myeloid leukemia, is also mainly mediated by CYP2C8, for which the CYP2C8*2 and CYP2C8*3 have a gain-of-function effect on imatinib while CYP2C8*4 polymorphism was opposite.335,336 In addition to influencing the pharmacokinetics and pharmacodynamics of those anticancer drugs, the question of whether CYP2C8 polymorphism affects the occurrence and development of tumors is also very important. The CYP2C8 genotype (rs1058930), those who have the CG allele, have a 7.74 degree increased risk of breast cancer (CI = 95% 0.95–62.5) in women in Mazandaran province.337 Moreover, the CYP2C8/9 *3/*1/*2/*1 genotype seem to be at higher risk of breast cancer recurrence (in tumors larger than 20 mm), especially in women treated with tamoxifen.338 However, the most common functional genetic variant i.e. CYP2C8*3 does not show a major association with colorectal cancer risk.339 CYP2C8*3 also demonstrated an impaired metabolism of AA to 11,12- and 14,15-EET, which were 26-45% that of wild-type CYP2C8*1.333
CYP2C9: CYP2C9 accounts for about 20% of hepatic CYP content and metabolizes about 10% of therapeutically relevant drugs such as the anticoagulant warfarin, the anticonvulsant phenytoin, the antidiabetic drug tolbutamide, and numerous NSAIDs.340,341 CYP2C9 also is involved in the bioactivation of several carcinogens such as polycyclic aromatic hydrocarbons (PAHs) and heterocyclic aromatic amines,342,343 and the generation of endogenous active substances, especially EETs,344 leading to be associated with cancer risk. Over 30 CYP2C9 alleles have been detected [http://www.cypalleles.ki.se/cyp2c9.htm]. Among them, the CYP2C9*2 (R144C) and CYP2C9*3 (I359L) variants occur at a high frequency among Caucasians with frequencies of 0.08–0.14 and 0.04–0.16, respectively,345,346,347 both yielding enzymes with decreased activity.347 Other variant CYP2C9 alleles with relatively low frequencies have also been reported, although no association studies between them and human cancer risk have been performed. An increased frequency of the CYP2C9*2 allele in patients with lung cancer has been found and was linked with an increased risk of lung carcinogenesis in a North-American population.348,349 Concerning colorectal cancer, individuals carrying CYP2C9*2 or *3 alleles are at increased risk of developing colorectal cancer possibly for CYP2C9-mediated metabolic activation of PAHs and heterocyclic aromatic amines and diminishing the protective effects of NSAIDs.350,351 In addition, a higher prevalence of cases with variant genotypes of CYP2C9*2 or *3 were associated with an increased risk to develop head and neck squamous cell carcinoma (HNSCC).352 Conversely, other studies found that CYP2C9 polymorphism did not show any association with the risk of lung or colorectal cancer in a Spanish population,353,354 and even decreased the risk of bladder cancer in a single case-control study with 958 cases and 1029 controls.355 Except for their actions on the risk of various cancers, functional variants in the CYP2C9 also altered the clinical impacts of anticancer drugs. For example, CYP2C9*2 heterozygotes increased the risk of an insufficient response to breast cancer neoadjuvant chemotherapy 4.64-fold higher (OR = 4.64, p = 0.02) than in patients with the wild-type allele.356 In addition, CYP2C9*2 and CYP2C9*3 metabolize AA less efficiently than CYP2C9*1 and that they play a role in the progression of non-small cell lung cancer (NSCLC) via impaired EET biosynthesis.357 Together, CYP epoxygenases (especially CYP2J2, CYP2C8, and CYP2C9) and AA-derived EETs were widely distributed in various tumors and play important roles in the initiation and development of cancer. Moreover, CYP polymorphisms are also closely associated with cancer fate.
CYP-derived HETEs and cancer
20-HETE has long been implicated in the proliferation of tumor cells and endothelial cells, often invoking the participation of growth factors, such as VEGF, epidermal growth factor (EGF), fibroblast growth factor (FGF), or platelet-derived growth factor (PDGF).358 Modulation of the CYP4:20-HETE pathway has very pronounced effects on tumor size in animal models of the brain, kidney, and breast cancer. For example, following implantation into normal rat forebrain of U251 glioma cells with CYP4A1 overexpression, a 10-fold increase in tumor volume was observed compared with the nontransfected cells.359 Similarly, chronic treatment with HET0016, a potent and selective CYP4 inhibitor, increased survival time by 5 days in 9L gliosarcoma tumors, apparently through a combination of reduced mitosis and increased apoptosis.360 Injection of mice with an NSCLC-derived cell line (A549) transfected with CYP4A11 increased the tumor size and growth rate, both of which were reversed by HET0016 or WIT002.361 In addition, several groups, applying independent experimental approaches, established a role for 20-HETE in angiogenesis in the early 2000s. In one of the first of these studies, angiogenesis induced in skeletal muscle by chronic electrical stimulation was accompanied by a 2.5-fold increase in a 20-HETE formation that could be completely blocked by HET0016.362 More recently, CYP4Z1 overexpression in breast cancer cells has been linked to increased VEGF expression, angiogenesis, cell proliferation, and migration in vitro as well as increased tumor weight in xenograft models.363 Recently, Zeldin and coworkers found that endothelial cells from CYP4F2 transgenic mouse exhibited a twofold increase in levels of 20-HETE, increased growth and tube formation with upregulation of VEGF and the prooxidant enzyme NADPH oxidase subunits (gp91phox and p47phox).364 In addition, endothelial progenitor cells (EPCs) express relatively high levels of CYP4A11 and 20-HETE,365 and in return 20-HETE has been shown to promote EPC angiogenesis both in vitro and in vivo.365,366 Collectively, these studies provide strong support for the CYP4:20-HETE pathway as a potential drug target for combating tumor growth and metastasis.
AA cascade and cancer-associated signaling pathways
Crosstalk between AA pathway associated enzymes and their metabolites regulate many pathophysiological processes in cellular systems and within the TME.367,368 The main biological functions of AA metabolites in cancer cells were concluded in Table 3.
AA-COXs, -LOXs and cancer
Several cytokines, including pro-inflammatory cytokines, induce the expression of cytolic PLA2, COX-2, and 5-LOX genes through activation of the IκB kinase (IKK)/IκB/NF-κB/AP-1/p300 pathway.369,370,371,372 TNFα induced cPLA2, COX-2, and 5-LOX expression were shown to be mediated by TNFR/p42/p44 MAPK/Elk-1/p300 and p38 MAPK- and JNK1/2-dependent AP-1/p300 pathways in human lung epithelial cells.369,371 IL-1β also elicits similar effects. Another cytokine, i.e., IL-8 activates cPLA2 via MAPK signaling pathway in PMNLs.8,373 Eicosanoids (PGs and LTs) stimulate the expressions of cPLA2, COX-2, and 5-LOX genes via activation of G-protein-coupled receptors (GPCR) mediated MAPK/NF-κB signaling pathways in cancer or pro-tumorigenic cells.374,375,376,377,378 Eicosanoids (PGs and LTs) can stimulate or promote tumor epithelial cell survival, proliferation, invasion, and metastasis and inhibit apoptosis by modulating multiple signaling pathways.1 Ultraviolet (UV) irradiation from solar exposure is a risk factor for carcinogenesis, which activates AA pathway via MAPK and NF-κB/AP-1-mediated signaling pathway. Yan et al.379 reported that UVB-induced LTB4 production and 5-LOX expression. COX-2 pharmacologic inhibition and COX-2 gene knockout prevent UVB-induced SKH-1 mouse skin tumorigenesis.8,380,381 Chen et al. found that UVB-induces the expression of cPLA2, which is mediated by oxidative stress. Black et al.382 reported that UVB-upregulated expression of COX-2 and other enzymes involved in PGs synthesis and TXA2, and 5-LOX and other enzymes involved in LTs synthesis, along with pro-inflammatory cytokines, namely IFNγ, IL-1β, TGF-β, and TNF-α in human corneal epithelial cells. The same investigators also found that inhibition of p38 MAPK blocked UVB-induced expression of COX-2, 15-LOX-2, and TNF-α, which demonstrate that UVB induces expression of COX-2, LOXs, and cytokines like TNF-α via MAPK signaling pathway.382 UVA upregulates the expression of COX-2 gene through MAPK/AP-1 mediated pathways.383 TPA/PMA, a tumor promoter, induces expression of COX-2 by activating MAPK/NF-κB/AP-1 mediated pathways whereas COX-2 inhibitors suppress.8,384,385 In addition, crystalline silica can promote lung tumor growth mediated by LTB4/BLT-1.386
Mechanisms of AA-CYP-EETs/sEH on cancer
In 2005, our laboratory explored the potential roles of CYP2J2 and its active products EETs on the neoplastic phenotype of carcinoma cells for the first time.261 Overexpression of CYP2J2 or addition of EETs to cultured carcinoma cell lines in vitro markedly accelerated proliferation, cell counts, cell cycle, and protected carcinoma cells from apoptosis induced by TNF-α. At the molecular levels, this involved the phosphorylation of EGFR and activation of PI3K/AKT and the MAPK signaling pathway. In contrast, either the downregulation of CYP2J2 transfection or the addition of epoxygenase inhibitors inhibited proliferation and accelerated TNF-α-induced apoptosis. In addition, carcinoma cells overexpressing CYP2J2 generated tumors at a faster rate and resulted in larger tumors than those generated from control carcinoma cells in vivo xenograft tumor model.261 Similarly, EETs promoted proliferation and increased the number of cells in the S/G2-M phase in a dose- and time-dependent manner in four tumor cell lines. The later effects were abolished by the inhibition of PI3K, MAPKK, MAPK, and PKC.387 Moreover, a specific inhibitor of CYP2J2 decreased EET production by ~60%, and inhibited the proliferation of human tumor cells at the same time as increasing tumor cell apoptosis via a caspase-3, Bcl-2 and Bax-dependent mechanism.388 Addition of exogenous EET or CYP2J2 overexpression also markedly accelerated proliferation and attenuated apoptosis in cultured human-derived malignant hematologic cell lines, which could be blocked by the pretreatment with the CYP2J2 inhibitor.314 Similar pro-proliferative and anti-apoptosic effects of the EETs were also observed in pheochromocytoma/paraganglioma tumors.389
CYP2J2 and CYP2C9 expression has also been correlated to high Ki-67 labeling indices in adenocarcinoma (EAC) and squamous cell carcinoma (ESCC). Selective inhibition of CYP2C9 decreased tumor cell proliferation and led to a G0/G1 phase cell-cycle arrest in vitro, which was abolished by the addition of 11,12-EET.388,390 Moreover, CYP3A4 is a highly active AA epoxygenase and synthesized AA epoxygenase products 8,9-, 11,12-, and 14,15-EET in the breast cancer lines.391 CYP3A4 silencing blocked the cell cycle at the G2/M checkpoint and induced apoptosis in the MCF7 line via inhibiting Stat3 (Tyr-705) phosphorylation, thereby inhibiting anchorage-dependent growth and survival. Knockdown of CYP3A5 and -2C8, both of which exhibit homology with CYP3A4, inhibited the proliferation of the MCF7, T47D, and MDA-MB-231 lines to varying degrees.391 Also, overexpression of CYP3A4 promoted the cell growth and cell cycle progression from the G1 to the S phase in a human hepatoma cell line, which was attenuated by a putative EET receptor antagonist, 14,15-EEZE and a PI3K inhibitor.392 These results suggest that CYP3A4 activity can accelerate tumor progression, which is independent of the activation of carcinogens and metabolism of anti-cancer drugs.
EETs and CYP2J2 transgenic mice attenuate doxorubicin-mediated cardiac damage by protecting mitochondria.151,393 More recently, 11,12-EET was reported to increase the expression of the antioxidant enzymes superoxide dismutase and catalase, and to attenuate mitochondrial transmembrane potential collapse and caspase activation in Tca-8113 cancer cells induced by the anti-leukemia drug arsenic trioxide.394 In addition, stably overexpressed CYP2J2 in a breast cancer cell line reduced the production of reactive oxygen species (ROS), thereby preventing cell death from anti-cancer agents such as paclitaxel, doxorubicin, sorafenib, and staurosporine.395 The expression and activity of aldehyde dehydrogenase 1A1 (ALDH1A1) were strongly upregulated in the CYP2J2 expressing cells and ALDH1A1 gene silencing restored their sensitivity to paclitaxel.395 CYP3A4 was found to be required for tumor formation in ER+/HER2− breast cancer by suppressing autophagy, in part, by inhibiting AMPK activation. The effect was also associated with mitochondria, where CYP3A4 promoted the activity of the electron transport chain and increased oxidative phosphorylation.396 CYP3A4 knockdown or inhibition by biguanides activated AMPKα, promoted autophagy, and prevented mammary tumor formation.397 These results indicate that AA metabolizing CYP epoxygenases and EETs also are associated with mitochondrial function and oxidative stress of cancer cells, which may be another potential mechanism of their anti-apoptosic actions.
Primary tumor formation is a necessary requirement for metastasis, and it is estimated that ~1 × 106 cells per gram of primary tumor escape into circulation per day. However, only a fraction of cells leaving the primary tumor to survive in circulation and even fewer cells colonize secondary sites.398 Jiang and his colleagues indicated that the overexpression of CYP2J2 or the exogenous application of EETs significantly induced tumor cell migration, invasion, adhesion to fibronectin, as well as colony-forming capacity.262 Consistently, CYP2J2 overexpression also enhanced metastatic potential in vivo and rAAV CYP2J2-infected human breast carcinoma cells developed 60% more lung metastases in athymic BALB/c mice.262 Selective inhibition of CYP2J2 prevented tumor cell adherence, invasion, and migration by decreasing the activation of the EGFR and PI3K/AKT pathways in vivo.388 Actin-myosin microfilament formation is closely associated with the invasion and migration of cancer cells. 11,12-EET was found to induced prostate carcinoma cell spreading and the formation of actin-myosin microfilaments possibly by the trans-activation of EGFR and PI3K/AKT pathways, which could account for the observed effects on cell invasion and migration.399 Blocking EET synthesis or activation using EET antagonists such as 14,15-EEZE, on the other hand, caused the cells to become more rounded and smaller.399 Together, these data suggest that CYP inhibition may represent a novel approach to prevent metastasis of human cancers. In addition, endothelium-derived EETs also contribute to tumor metastasis. Briefly, endothelial-specific expression of either CYP2C8 or CYP2J2 (Tie2-CYP2C8-Tr, Tie2-CYP2J2-Tr) accelerated the escape from tumor dormancy and extensive multi-organ metastasis.400
The TME is composed of several distinct cell types, including fibroblasts, pericytes, immune cells, adipocytes, endothelial cells, and a noncellular compartment, the extracellular matrix. The cross talk between cancer and stromal cells in the TME promotes does much to create optimal conditions to support cancer cell growth, invasion, angiogenesis, and metastasis. These stromal cells have also been recognized as attractive targets to reduce resistance to anticancer therapy and tumor recurrence.401,402
Inflammatory mediators and inflammatory cells in the inflammatory microenvironment promote the transformation of normal cells to cancer cells in the early stage of cancer, promote the growth and development of cancer cells, and induce tumor immune escape.367 An early paper demonstrated that physiological concentrations of EETs or overexpression of CYP2J2 prevented leukocyte adhesion to the vascular wall by a mechanism involving inhibition of transcription factor NF-κB and IκB kinase.403,404 Similarly, CYP2J2 transgenic, CYP2C8 transgenic and sEH-/- mice each exhibited a significant attenuation of endotoxin-induced activation of NF-κB signaling, cellular adhesion molecule, chemokine, and cytokine expression, and neutrophil infiltration in vivo.202 That is, inhibition of NF-κB is one of the central mediators of the anti-inflammatory response of EETs. NF-κB had been generally recognized as a critical link between chronic inflammation and cancer.405 Thus, it is tempting to speculate that the CYP-EET/sEH system in TME could manipulate the activation state of immune cells thus contribution to tumor suppression. However, many CYP enzymes, e.g., CYP2C8 and 9 generate reactive oxygen species as a byproduct of their reaction which can, in turn, stimulate NF-κB. In the vascular system this has been linked with an increased adhesion molecular expression and detrimental effects on vascular function.406 Thus, the actions of CYP enzymes on the NF-κB pathway seem to depend on their ability to generate biologically relevant amounts of oxygen-derived free radicals (e.g., CYP2C8 and CYP2C9) while others (e.g., CYP2J2) generate fewer such mediators (Fig. 3).

The mechanisms of actions of EETs on tumor growth. EETs accelerated proliferation, cell cycle, and protected carcinoma cells from apoptosis through multiple signal transduction pathways. Besides, EETs improved mitochondrial function and prevented carcinoma cells from oxidative stress damage. In addition, EETs also were found to regulate multiple important cells in TME, such as promoting endothelial cell angiogenesis, fibroblast activation, and anti-inflammation
EETs also increase PPARγ transcription and blocking PPARγ reduces the antiinflammatory effects of the EETs and sEH inhibitors, indicating PPARγ is an effector of EETs.407 The infiltrating tumor-associated macrophages are an important additional source of VEGFA, leading to increased vascular permeability and tumor cell metastasis in most solid tumors.408 Some monocyte CYP enzymes are differentially expressed in tumor macrophages, one example being CYP2S1 which could be detected in inflamed tissues but was lacking from tumor-associated macrophages in breast cancer metastases.409 In addition, CYP4A positive tumor-associated macrophages correlated positively with metastatic niche formation and poor outcome in breast cancer therapy. The inhibition of CYP4A, on the other hand, tended to reduce pre-metastatic niche formation, reflected in a reduced VEGFR-1 positive myeloid cell recruitment.410 Apart from macrophages, infiltrating neutrophils also stimulate angiogenesis by secreting VEGFA and other angiogenic growth factors. 14,15-EET was found to induce neutrophil infiltration into dormant metastases to induce a neutrophil reprogramming from a tumor-suppressing to a tumor-promoting phenotype. Depleting neutrophils resulted in the failure of 14,15-EET to promote the development of micro-metastases.411
The Hammock laboratory demonstrated that the sEH is a therapeutic target for inflammation for its capacity of inactivating endogenous anti-inflammatory EETs.412 In another study, the tobacco smoke-exposed rats treated with a sEH inhibitor resulted in a significant decrease in bronchoalveolar inflammatory cells, including significant reductions in neutrophils, alveolar macrophages, and lymphocytes.412,413 sEH inhibition decreases COX-2 protein levels without altering COX-1 expression and decreases inflammatory eicosanoid levels in LPS-challenged mice. The inhibitors also showed synergetic effects with NSAIDs and COX-2 inhibitors in suppressing inflammation.414 Thus, sEH inhibitors could be a novel therapeutic method for inflammation-related cancer via their strong anti-inflammation actions especially when combined with COX inhibition, although the pro-angiogenic and tumorigenic products EETs increase.
AA metabolism in other inflammatory and metabolic diseases
AA in asthma
Asthma is a chronic disease of the lung caused by airway inflammation and involves airway hyperresponsiveness, mucus overproduction, remodeling, and narrowing of the airway. CysLT1 antagonists, including montelukast, pranlukast, and zafirlukast, are used worldwide for the treatment of asthma. Asthma is mostly associated with type 2 inflammation (The type 2 inflammation is mainly regulated by subpopulations of CD4+ T cells known as T helper 2 cells), leading to the release of Th2 cytokines (IL-4, IL-5, and IL-13), IgE production, airway recruitment of eosinophils, and goblet cell metaplasia. It is now understood that some asthmatic inflammation induces the recruitment and activation of Th2 cells and group 2 innate lymphoid cells (ILC2s) by epithelial-derived innate cytokines such as IL-25, IL-33, and thymic stromal lymphopoietin (TSLP).415,416 A recent study demonstrated that IL-33 enhanced CysLT1 expression in human peripheral blood lymphocytes (PBLs) in vitro.417 LTD4 stimulation induces intracellular calcium mobilization and chemotaxis in PBLs, which express a membrane-bound IL-33-specific receptor, ST2L. Boyce et al. and Doherty et al. independently demonstrated that LTC4 potentiated the activation and migration of ILC2s via CysLT1 or CysLT2 signaling.304,418,419 Thus, the CysLT antagonists suppress innate immunological function in asthmatic patients. In addition, LTE4 induces mucin release and submucosal swelling in the nasal mucosa through GPR99 signaling in lung and nasal epithelial cells.420 LTE4 stimulation greatly facilitated the chemotaxis of ILC2s differentiated from human PBLs.421 LTE4 also enhances the release of Th2 cytokines and pro-inflammatory cytokines (e.g., IL-8 and GM-CSF) from cultured ILC2s in combination with PGD2, IL-25, IL-33, and TSLP.421 Thus, in addition to CysLT1 and CysLT2, the LTE4 receptor GPR99 may be a useful therapeutic target for asthma and related diseases such as aspirin-exacerbated respiratory disease.121,422
AA in arthritis
Arthritis is a common inflammatory disease of the joints and includes rheumatoid arthritis (RA) and osteoarthritis. RA is a chronic and systemic inflammatory autoimmune disease that is characterized by inflammatory cell infiltration, synovial hyperplasia, and bone and cartilage destruction.423 There are both acute and chronic models of rheumatoid arthritis, the mice expressing both the T cell receptor (TCR) transgene KRN and the MHC class II molecule Ag7 (K/BxN mice) and collagen-induced arthritis (CIA) model, respectively.424
NSAIDs are predominantly used for controlling pain and inflammation and are administered as a first‐line medication for newly diagnosed RA cases. Recently, researchers have concentrated mostly on NSAIDs that inhibit COX‐2 selectively. Their pain alleviating and anti‐inflammatory effects are similar to conventional NSAIDs.425,426
Luster and Haribabu et al. clearly demonstrate that BLT1 is critically involved in inflammatory arthritis using several RA mouse models, including the K/BxN STA304,427,428 and CIA429 models. In the K/BxN STA model, the authors demonstrated that BLT1 is required for peripheral neutrophil recruitment into the joint and the resultant induction of IL-1 via immune complex-Fc R interactions. Inflammatory cytokines such as IL-1β and TNFα accelerate the production of the chemokines CXCL1, CXCL5, and CCL5 from fibroblast-like synovial cells, endothelial cells, and macrophages. These chemokines subsequently promote the late phase of neutrophil recruitment by activation of CCR1 and CXCR2, the receptors for CCL5 and CXCL1/5, respectively. Importantly, a BLT1 antagonist, CP105696, improved the incidence of arthritis in both the preventive and therapeutic modes.304 Taken together, BLT1 may be a promising therapeutic target for arthritis.
AA cascades in homeostasis of metabolic diseases
Diabetes mellitus type 1 (DM1) and DM2 are by definition associated with recurrent hyperglycemia due to insufficient insulin production and insulin resistance, respectively. Hyperglycemia induces the production of pro-inflammatory mediators by PMNs, gives rise to oxygen radical formation, hampers PMN chemotaxis, and supports the adhesion of PMNs to the vasculature in diabetic mice.78,430 In addition, free fatty acids activate the NLRP3-ASC inflammasome, and a disruption of the associated Nod-like receptors (NLRs) protects against insulin resistance and hyperglycemia in obesity.431 Glucose and lipid metabolism share various metabolic pathways. Consequently, disturbances in glucose and lipid metabolism are tightly related, and over-nutrition and/or obesity ensure both, dysregulated lipid metabolism and hyperglycemia. Macrophages derived from diabetic mice have a pro-inflammatory phenotype and express high levels of acyl-CoA synthetase 1 (ACSL1). ACSL1 is implicated in the generation of pro-inflammatory PGs, such as PGE2, thus fostering pro-inflammatory functions of macrophages.432 Consequently, disruption of ACSL1 in myeloid cells significantly reduces the inflammatory signaling in diabetic macrophages and attenuates the progression of the atherosclerotic lesions in diabetic mice.432 PGE2 inhibits insulin secretion in pancreatic islets, and enhances pancreatic β cell dysfunction and destruction, whereas PGI2 improves the insulin sensitivity of pancreatic cells. Contrarily, PGE2 fosters adipogenesis in white fat tissue and induces glycogenolysis and gluconeogenesis, thus alleviating insulin resistance of adipocytes.433 Recently, PGF2, which is synthesized at higher levels in diabetic mice, was linked to hepatic gluconeogenesis, a major driver of fasting hyperglycemia in DM2.8,434
12/15-LOX enzymes are linked to DM via the production of various HPETEs, which interact with PPARs, and are implicated in the cytokine-mediated damage of pancreatic cells. It is therefore not surprising that 12/15-LOX knockout mice demonstrate a partial resistance to diabetes development.433,435,436,437 Similarly, LTs, produced by 5-LOX and 12-LOX-derived HETEs, inhibits pancreatic insulin secretion, and the genetic disruption or pharmacological inhibition of these LOXs protects against pancreatic islet cell destruction in diabetic mice.438 LTB4 has been found to be essential for the recruitment and activation of adipose tissue B2 lymphocytes, which contribute to the establishment of insulin resistance following a high-fat diet.439
CYP-derived EETs and 20-HETE induce insulin secretion and protect pancreatic islet cells from apoptosis.21,440 Diabetes and obesity are associated with an enhanced expression of the sEH, and genetic deletion of the sEH ensues an improved insulin sensitivity and an anti-apoptotic effect on pancreas islet cells in the murine diabetes model.441 Recent data suggest that CYP enzymes and EETs are involved in the homeostasis of metabolic diseases, including obesity and diabetes.442,443 Previous study has also shown that sEH is expressed in adipose tissue,444 hepatocytes,445, and pancreatic islets. At least in part, it is speculated that EETs play an important role in the treatment of diet-associated metabolic diseases. Our previous study indicated that, in addition to lowering blood pressure, CYP2J3 overexpression improved insulin resistance in rats treated with fructose and in db/db diabetic mice. This improvement in insulin resistance was associated with the activation of insulin receptor signaling and adiponectin-mediated AMPK signaling pathways.442,446 CYP2J3 gene delivery markedly reversed insulin resistance via upregulated AMPK signaling, which was associated with decreased ER stress response in adipose tissue.442 CYP2J3-derived EETs alleviate insulin resistance, at least in part through upregulated endothelial nitric oxide synthase expression in rats treated with fructose, which was associated with activation of MAPK and protein kinase C signaling pathways. Genetic disruption or pharmacologic inhibition of sEH led to an enhancement of insulin signaling and sensitivity, increased islet size and vasculature, and decreased plasma glucose.447 sEH knockout or inhibition not only attenuated insulin resistance in diabetes but also enhanced glucose-stimulated insulin secretion from islet cells and decreased islet cell apoptosis. Interestingly, several studies have shown that the disruption of sEH enhanced islet glucose-stimulated insulin secretion through AMPK signaling and decreased islet cell apoptosis in diabetes.447 Inhibiting sEH activity provided significant protection against islet β cell damage and improved glucose homeostasis in streptozotocin-induced diabetes.447,448 Moreover, 5,6-EET directly stimulates the release of insulin but has no effect on glucagon release. In contrast, 8,9-EET, 11,12-EET, and 14,15-EET increase glucagon release without affecting insulin secretion.449 Accordingly, the therapeutic potential of sEH inhibitors was tested in several clinical trials. Whereas results of some trials are still pending (e.g., NCT03486223), a Phase II trial introducing a thrice-daily application of an orally administered sEH inhibitor in patients with mild to moderate arterial hypertension and pre-diabetes, failed to demonstrate an improvement of insulin sensitivity (NCT00847899).
Finally, AA also facilitates the production of anti-inflammatory LXs. The latter was reported to improve insulin sensitivity and may prevent the development of DM.450 For instance, LXA4 inhibits IL-6, TNFα, and ROS production thus hampers obesity-associated inflammation and has an anti-diabetic effect.451,452,453 LXs are endogenously produced eicosanoids with a spectrum of anti-inflammatory, proresolution, and antifibrotic bioactions. Furthermore, LXs stimulate nonphlogistic macrophage phagocytosis of apoptotic neutrophils both in vitro and in vivo, which is also associated with a shift from the release of proinflammatory to anti-inflammatory cytokines.451,454
Adipose tissue is a metabolically active endocrine organ, comprising adipocytes and other cells, such as macrophages and preadipocytes. A key factor in the development of adipose inflammation is a switch in the phenotype of the adipose tissue macrophages (ATMs). Tissue macrophages are heterogeneous and display phenotypic plasticity. Classically activated M1 macrophages are described as proinflammatory, whereas alternatively activated M2 macrophages are thought to be proresolving.451,455 In lean subjects, ATMs are predominantly of an M2 phenotype. However, factors such as obesity cause adipose hypoxia and hyperglycemia, the latter, e.g., leading to steatosis and hepatic stress responses with the production of proinflammatory mediators, contributing to systemic inflammation. In combination, these factors cause adipose inflammation and recruitment of macrophages, predominantly of an M1 phenotype.451,456 M1 ATMs secrete proinflammatory mediators, which further exaggerate inflammatory responses promoting adipose insulin resistance. The subsequent release of free fatty acids results in systemic lipotoxicity, which contributes to the pathology of T2DM. It has previously been shown that macrophage depletion or blocking macrophage recruitment protects mice from adipose inflammation and IR.457 Promoting a shift of M1 to M2 phenotype may, however, be a more physiological approach to subverting adipose inflammation, since the macrophages are required for effective resolution. A previous study reported that LXA4 treatment of macrophages subverted macrophage-induced IR and restored glucose uptake in adipocytes. This effect was associated with rescued AKT activation and reduced secretion of proinflammatory cytokines, including TNFα. These data expand the repertoire of bioactions associated with LXA4 and provide initial ex vivo and in vitro evidence to support the potential of using proresolving mediators, such as LXA4, as a therapeutic to reduce adipose inflammation and IR for instance in T2DM.451
In summary, AA derivatives play diverse and partly contrasting roles in the pathogenesis of DM. Therefore, research in AA metabolism and its enzymatic pathways may identify novel targets for the treatment of DM and its associated co-morbidities.
Clinical studies by targeting AA pathway and outlook for novel therapeutic applications
Based on the widely established concept that COX, LOX, and the CYP-EET/sEH axis play important roles in cardiovascular disease as well as in tumor growth and metastasis, the development of drugs or biological products that target COX, LOX, CYP enzymes, or the sEH has bright prospects.
Clinical trials associated with AA-COX pathway
Since bioactive lipid metabolites from AA metabolism can be potent mediators of inflammation and cancer progression, COX inhibitors act as important mediators of these cellular responses. As known, aspirin acts as a drug against pain and inflammation, has been widely used in many solid cancers, such as lung cancer, colorectal cancer, and esophageal cancer (e.g., NCT02169271, NCT00468910, NCT00474903). In addition, overexpression of COX-2 in several different types of solid tumors has been reported and supported by animal studies that confirmed the association of genetic COX-2 overexpression with tumorigenesis and malignant progression.458 Consistently, more and more s clinical trials showed that COX-2 may be an important target in cancer therapies. Various COX-2 inhibitors (e.g., celecoxib, apricoxib) have also been developed against different cancers (e.g., NCT00582660, NCT00466505, NCT01111591, NCT01532362). In addition to cancers, aspirin also has been used as a medicament for antiplatelet aggregation. It is often prescribed by coronary artery disease patients because of its unique ability to permanently prevent platelets from aggregating and forming a blood clot. In addition, more and more COX-2 inhibitors (such as naproxen sodium, etoricoxib and celecoxib) have been introduced in preventing pain and inflammation in arthritis and osteoarthritis (e.g., NCT03699293, NCT00746720, NCT02198924). Specially, the PGI2 derivative, beraprost, has been reported to reduce pulmonary arterial hypertension (PAH) (NCT00990314). Moreover, treprostinil, a DP1 and EP2 agonist and selexipag, an IP receptor agonist, were both newly approved by FDA to treat PAH (e.g., NCT01268553, NCT01106014) (Table 4).
Clinical trials associated with AA-LOX pathway
Recent studies showed that a 5-LOX inhibitor, VIA-2291, possessed a protective role against coronary artery disease (NCT00358826) and atherosclerosis (NCT00352417). Recently, another specific 5-LOX inhibitor, zileuton, usually used to modify airway inflammation (NCT00723021), was also found to prevent tumor growth (NCT00056004 and NCT02047149). Montelukast, an effective drug against asthma, was also found to prevent coronary artery disease by targeting Cys-LT1-receptor (NCT00379808). Besides, in obesity or T2DM, montelukast may have a role in regulating homeostasis of metabolic diseases (NCT04075110) (Table 4). Despite these promising effects in both asthma and in CVD montelukast may lead to severe neurospyschiatric problems. The biologic mechanisms underlying the neuropsychiatric events are not well understood, but evidence from animal studies suggests that montelukast could act directly on cells in the brain. Orally administered montelukast (10 mg/kg/day, 7 days) was detectable in brain tissue and cerebrospinal fluid (CSF) in rats,459 providing evidence for its ability to cross the blood-brain barrier. Montelukast is a potent competitive antagonist (IC50 = 2.3 nM) at its target, the CysLT1 receptor.460 However, expression of the CysLT1R in the normal human brain is very low/non-existent, implying that the compound may have off-tartet effets Montelukast is also a competitive antagonist of (IC50 = ~60 nM) of GPR17, a G-protein-coupled receptor, which is expressed on neurons and glial cells in the human brain.461 GPR17 is recognized as a regulator of oligodendrocyte development and remyelinating function.462 Montelukast inhibition of GPR17 function on neurons and/or glial cells may contribute to the biologic processes underlying the observed neuropsychiatric events associated with montelukast treatment.
CYP epoxygenases inhibitors and EET antagonists
There are no clinical trials that directly targeting CYP enzymes or their direct products. Thus, below, we will mainly introduce the CYP inhibitors and EET antagonists, which may have the potential to be used in the future.
Both CYP epoxygenases inhibitors and EET antagonists are effective approaches to reduce EETs production and their biological effects. Two fatty acid derivatives [2-(2-propynyloxy)-benzenehexanoic acid (PPOH) and its metabolically stable congener N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH)] are generally used as specific EETS synthesis inhibitors.463 The former compound shows wide inhibition on CYP2B and 2C epoxygenases while MS-PPOH prefers to inhibit CYP2C9 and CYP2C11 subtypes.464 The lipid-lowering drug gemfibrozil also shows widespread inhibition on CYP epoxygenases including CYP2C8 with a Ki range between 9.3 and 270 mM, CYP2C9 and CYP2C19 with Ki values of 5.8 and 24 mM, respectively, and CYP1A2 with a Ki of 82 mM.465 In vitro study, MS-PPOH abolished migration and tube formation of endothelial cells exposed to hypoxia or CYP2C9 overexpression. In addition, blocking EET synthesis by MS-PPOH] impaired the ability of prostate carcinoma cells (PC-3, DU-145, and LNCaP) to invade and migrate.399 In both primary and secondary prevention studies, gemfibrozil reduced cardiovascular endpoints and coronary disease mortality.466,467 A number of recent studies reveal that apart from its lipid-lowering effects, gemfibrozil can also regulate many other signaling pathways responsible for inflammation, switching of T-helper cells, cell-to-cell contact, migration, and oxidative stress.468 In addition, another epoxygenase inhibitor 17-ODYA had also been found to inhibit the proliferation, migration, invasion, and adhesion in solid cancer cells262 and multiple myeloma cells,469 and accelerate cancer cell apoptosis induced by TNFα.261 Human umbilical vein endothelial cell proliferation and tube formation are also restrained by 17-ODYA treatment with an associated reduction in EET production.212,470 In addition, CYP3A4, another epoxygenase responsible for EET production, was highly expressed in breast cancer and associated with breast cancer development and progression.471 Treatment of breast cancer cells with ketoconazole and azamulin, selective inhibitors of CYP3A4, inhibited cell proliferation and conferred sensitivity to the selective estrogen receptor modulator 4-hydroxytamoxifen.472 Thus, CYP epoxygenases inhibitors are expected to be potential drugs against tumor growth and metastasis via endothelium-dependent and independent mechanisms. Although various CYP epoxygenases inhibitors (e.g., SKF525A, clotrimazole) had been synthesized,473 few inhibitors enter into clinical trials as an anticancer therapy.474 Firstly, these inhibitors often target multiple CYP homologous genes, resulting in changes of various lipid metabolites. Secondly, inhibiting CYP epoxygenase pathway may be followed by an increase of other arms of the eicosanoid pathways, such as COX or LOX activity, resulting in the generation of metabolites with angiogenic and tumorigenic potential. In addition, CYP inhibition influenced the bioavailability of anticancer agents such as paclitaxel and docetaxel, vinorelbine, and tamoxifen,475 limiting their clinical promotion.
As for EET-receptor antagonist, it’s a pity that so far, the EET receptor has not been conclusively identified despite numerous evidence linking the presumptive receptor to a GPCR. Therefore, developing compounds specifically binding to the uncertain EET receptor seems to be impractical. Interestingly, the synthetic 14,15-EET analogues, such as 14,15-EEZE, 14,15-epoxyeicosa-5(Z)-enoic acid 2-[2-(3-hydroxypropoxy)-ethoxy]-ethyl ester [14,15-EEZE-PEG] and 14,15-epoxyeicosa-5(Z)-enoic-methylsulfonylimide [14,15-EEZE-mSI], competitively suppressed the effects of EETs and are identified as EET-specific antagonists.399 Cancer cells treated with synthetic EET antagonists prevented EET-induced cell invasion and migration in vitro.399 In addition, 14,15-EEZE significantly inhibited migration392 and proliferation of CYP3A4 enhanced tumor cells and endothelial cells overexpressing CYP2C9.472 Consistent with these in vitro findings, mice treated with EET antagonists showed reduced primary tumor growth and multi-organ metastatic potential.400
The sEH inhibitors, stabilizing endogenous EETs, are promising drug candidates for multiple human diseases. In prophase animal models, various pharmacological sEHIs, such as AUDA, AUDA-BE, t-AUCB, TPPU, and 1-adamantan-1-yl-3-urea (AEPU), showed that they are able to effectively lower hypertension,476 alleviate multi-organ inflammation477,478 and neuropathic pain,413,479 inhibit cardiac hypertrophy,480 detrimental cardiac remodelings and HF,176 as well as to attenuate hepatocellular necrosis and hepatic fibrosis481 and renal interstitial fibrosis and inflammation.482
Given the pro-angiogenic and pro-tumorigenic action of EETs, reduction of EET synthesis may provide clinical benefit for cancer patients. Many researchers emphasized that the anti-tumor and anti-metastatic roles of PPARα activation depended on the suppression of endothelial function.483,484,485 A study conducted by Pozzi et al.486 pointed out that the anti-tumorigenic and anti-angiogenic properties of PPARα are AA epoxygenase-mediated. Treatment with PPARα ligands such as Wy-14643 or fibrates downregulates CYP2C9 and CYP2C44 expression in human and murine endothelial cells, respectively, and reduces EET biosynthesis.484,486 In a mouse xenograft model of tumorigenesis, disruption of host CYP2C44 epoxygenase suppressed tumor growth and vascularization and abrogated the anti-tumor effects of PPARα agonists.486 In addition, Mice treated with PPARα ligands also show reduced primary and metastatic non-small cell lung cancer (NSCLC) tumor growth, tumor angiogenesis, endothelial CYP2C44 expression, and circulating EET levels.487 Taken together, these results indicate that activation of PPARα and consequent downregulation of CYP2C expression may be a promising anti-cancer approach.
Besides PPAR, other nuclear receptors, including the aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), and glucocorticoid receptor (GR), were noted to participate in receptor-dependent mechanisms of CYP induction,488,489 where they directly bind to their response DNA sequences to regulate CYP gene expression. Genetic studies indicated that the AhR forms heterodimers with AhR nuclear translocator (ARNT), and then binds to xenobiotic response elements in promoter regions of CYP490 18686044. CAR targets and regulates CYP3A4, CYP2C8, and CYP2C9 in response to phenobarbital treatment.491,492 In human hepatocytes, PXR activates CYP3A genes in response to diverse chemicals, including certain natural and synthetic steroids, steroid metabolites, and several clinical drugs. In addition, PXR also activates other CYP genes including members of the 2B and 2C families.493 PXR, CAR, and PPAR are orphan receptors, which belong to the nuclear receptor/steroid receptor superfamily, play transcriptional regulatory roles via forming heterodimerize with the retinoid X receptor (RXR) after activation in the nucleus. Expression of PXR, CAR, and RXR are under transcriptional control of the GR. Therefore, the expression of CYP genes may be controlled by a cascade of signal transmissions: GR-[PXR/CAR/RXR]-CYP. In addition, hepatocyte nuclear factor 4α (HNF4α) and other members of liver-enriched transcription factors, including HNF1α, HNF2α, CCAAT/enhancer-binding protein α (C/EBPα), HNF3γ (FOXA), and HNF6, have been shown to regulate the constitutive expression of CYP2C genes.494,495 This extensive regulatory network provides the potential for the development of drugs targeted at inhibiting CYP-EETs.
MicroRNA-mediated regulation of CYP epoxygenases and tumor therapy
MicroRNAs (miRNA) are short non-coding RNA molecules of 21–23 nucleotides that modulate the stability and/or the translational efficiency of target messenger RNAs.496 Several miRNAs had been associated with the regulation of CYP epoxygenases function, which presents a novel and attractive avenue for cancer therapy.497 Chen et al.498 found the expression level of CYP2J2 was inversely proportional to that of let-7b in lung squamous cell cancer tissues and further uncovered that let-7b diminished cell proliferation and promoted apoptosis of tumor cells via posttranscriptional repression of CYP2J2. In addition, the upregulation of miR-128-3p is inversely correlated with the expression of CYP2C9 in hepatocellular carcinoma tissues. MiR-128-3p is able to suppress CYP2C9 expression/production in human hepatic cells by specifically targeting the 3’-UTR of CYP2C9 mRNA molecules.499 The expression of CYP2C9 is also reported to be directly and negatively regulated by miR-130b.500 The translation efficiency (protein/mRNA ratio) for CYP2C8 was significantly inhibited by miR-103 and miR-107, which also targeted CYP2C9 and CYP2C19 to a lesser degree than CYP2C8 in the human Liver.501 Taken together, miRNAs-mediated regulation of CYP epoxygenases may contribute to cancer treatment. Especially, various nanoparticles are being developed and employed to load microRNAs, overcoming challenges associated with microRNA degradation, transient expression and poor targeting.502
The potential risks of drugs targeting AA pathway for human application
Although drugs targeted at AA metabolism exhibited multiple therapeutic effects on CAD and cancer, their possible side effects deserve mention here. The gastrointestinal (GI) side effects are ranked as the most common among NSAIDs-related adverse events. However, COX-2 selective NSAID agents seem to reduce GI side effects compared with traditional non-selective drugs.503 In addition, another major concern across all forms of NSAID therapy is the cardiovascular side effects. Except for aspirin, other drugs in the NSAIDs class are associated with increased risk of cardiovascular side effects including hypertension, stroke, heart attacks, and HF.504 Up to now, zileuton is the only approved 5-LOX inhibitor but it has numerous disadvantages, such as hepatic toxicity and adverse pharmacokinetic profile derived from a short half-life.505 Masferrer et al.506, demonstrated the inhibitory potency of PF-4191834 on LTB4 production by use of rat air pouch model. PF-4191834 has also completed phase II (NCT00723021) clinical trial for asthma but phase II for knee osteoarthritis was terminated due to a serious adverse event (NCT01147458) such as syncope, acute hepatitis, and gastric ulcer hemorrhage. Little clinical data regarding the safety of 12/15 LOX inhibitors can be referenced, because their side effects usually preclude them from entering into routine clinical use.507 Antagonists against cysteinyl leukotriene receptor (CysLTR) type 1, including montelukast, pranlukast, and zafirlukast, has been linked to apparent liver injury, various neuropsychiatric events508 and skin adverse reactions.509 CYP inhibitors seem be well tolerated and tested with low risk. On the one hand, numerous substances in nature and many long-used drugs in clinical practice are non-selective inhibitors of CYP. On the other hand, the activation of the compensatory pathway makes it possible for another pathway to enhance compensation when one CYP enzyme is inhibited. For example, type 1 angiotensin-II receptor antagonist telmisartan used as an antihypertensive drug and H1 receptor antagonists, terfenadine used as antiallergic agent for many years, have been identified as potent CYP2J2 inhibitor at concentrations that are reached during clinical use and are well tolerated by patients,510,511 because terfenadone strongly inhibited CYP2J2-mediated metabolism process.512 However, the side effects of CYP inhibitors can not be ignored because of the important effects of CYP enzyme on drug metabolism and their double-edged sword on CVD and cancer treatment. No sEH inhibitor has been presented to the market yet. Only some hopeful sEH inhibitor candidates are subjected now to clinical trials such as GSK2256294A in Phase-I and AR9281 in Phase-II against chronic obstructive pulmonary disease (COPD) and hypertension.513,514 However, the possibility of angiogenic effects when inhibiting sEH needs to be further evaluated.
Conclusion and future direction
Considerable data indicate that COX, LOX, CYP enzymes, and their metabolites of AA play important roles in the initiation and development of human diseases, especially cardiovascular and cancer. Although the specific mechanisms are not entirely clear, increasing evidence indicates that the CYP pathway has potential as a therapeutic target in these two disease areas. An important challenge for future research will be to obtain a better understanding of the different biological activities of AA metabolites such as EETs generated by the CYP/sEH axis serve both endogenous cardiovascular protectors and promotor of cancers. Ultimately, understanding the basic cellular mechanisms of these metabolites will enhance our knowledge and lead to better management of CVD and cancer and well as inflammatory diseases via developing novel drugs in key point of AA metabolism pathways.
References
Wang, D. & Dubois, R. N. Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 (2010).
Bahia, M. S. et al. Inhibitors of microsomal prostaglandin E2 synthase-1 enzyme as emerging anti-inflammatory candidates. Med. Res. Rev. 34, 825–855 (2014).
Capra, V. et al. Eicosanoids and their drugs in cardiovascular diseases: focus on atherosclerosis and stroke. Med. Res. Rev. 33, 364–438 (2013).
Patrignani, P. & Patrono, C. Aspirin and cancer. J. Am. Coll. Cardiol. 68, 967–976 (2016).
Grosser, T., Ricciotti, E. & FitzGerald, G. A. The cardiovascular pharmacology of nonsteroidal anti-inflammatory drugs. Trends Pharmacol. Sci. 38, 733–748 (2017).
Sala, A., Proschak, E., Steinhilber, D. & Rovati, G. E. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem. Pharmacol. 158, 161–173 (2018).
Dubois, R. N. et al. Cyclooxygenase in biology and disease. FASEB J. 12, 1063–1073 (1998).
Yarla, N. S. et al. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin. Cancer Biol. 40-41, 48–81 (2016).
Fitzgerald, G. A. Coxibs and cardiovascular disease. N. Engl. J. Med. 351, 1709–1711 (2004).
Sharma, V. et al. Recent advancement in the discovery and development of COX-2 inhibitors: insight into biological activities and SAR studies (2008-2019). Bioorg. Chem. 89, 103007 (2019).
Yu, Y. et al. COX-2 modulates blood pressure and thrombosis in mice. Sci. Transl. Med. 4, 132ra154 (2012).
Mitchell, J. A. & Kirkby, N. S. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br. J. Pharmacol. 176, 1038–1050 (2019).
Steiropoulos, P., Trakada, G. & Bouros, D. Current pharmacological treatment of pulmonary arterial hypertension. Curr. Clin. Pharm. 3, 11–19 (2008).
Samuelsson, B. Prostaglandins, thromboxanes, and leukotrienes: formation and biological roles. Harvey Lect. 75, 1–40 (1979).
Yokomizo, T., Nakamura, M. & Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Investig. 128, 2691–2701 (2018).
Diamant, Z., Bjermer, M. E. & Montelukast, L. in the treatment of asthma and beyond. Expert Rev. Clin. Immunol. 5, 639–658 (2009).
Capdevila, J. et al. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proc. Natl Acad. Sci. USA 79, 767–770 (1982).
Tsai, M. J. et al. Montelukast induces apoptosis-inducing factor-mediated cell death of lung cancer cells. Int. J. Mol. Sci. 18, 1353 (2017).
Campbell, W. B., Gebremedhin, D., Pratt, P. F. & Harder, D. R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Res. 78, 415–423 (1996).
Zeldin, D. C. Epoxygenase pathways of arachidonic acid metabolism. J. Biol. Chem. 276, 36059–36062 (2001).
Xu, X., Zhang, X. A. & Wang, D. W. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv. Drug Deliv. Rev. 63, 597–609 (2011).
Imig, J. D. Prospective for cytochrome P450 epoxygenase cardiovascular and renal therapeutics. Pharmacol. Ther. 192, 1–19 (2018).
Panigrahy, D. et al. EET signaling in cancer. Cancer Metastasis Rev. 30, 525–540 (2011).
Wlodawer, P. & Samuelsson, B. On the organization and mechanism of prostaglandin synthetase. J. Biol. Chem. 248, 5673–5678 (1973).
Smith, W. L., DeWitt, D. L. & Garavito, R. M. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 (2000).
FitzGerald, G. A. & Patrono, C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 345, 433–442 (2001).
Liu, J. et al. Prostaglandin endoperoxide H synthases: peroxidase hydroperoxide specificity and cyclooxygenase activation. J. Biol. Chem. 282, 18233–18244 (2007).
Marnett, L. J. et al. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J. Biol. Chem. 274, 22903–22906 (1999).
Byrne, M. F. et al. pylori induces cyclooxygenase-1 and cyclooxygenase-2 expression in vascular endothelial cells. Scand. J. Gastroenterol. 38, 1023–1030 (2003).
Cullen, L., Kelly, L., Connor, S. O. & Fitzgerald, D. J. Selective cyclooxygenase-2 inhibition by nimesulide in man. J. Pharmacol. Exp. Ther. 287, 578–582 (1998).
Basu, S. Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Med. Res. Rev. 27, 435–468 (2007).
Brock, T. G., McNish, R. W. & Peters-Golden, M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem. 274, 11660–11666 (1999).
McAdam, B. F. et al. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc. Natl Acad. Sci. USA 96, 272–277 (1999).
Vara-Messler, M. et al. A potential role of PUFAs and COXIBs in cancer chemoprevention. Prostaglandins Other Lipid Mediators 120, 97–102 (2015).
Morita, I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediators 68-69, 165–175 (2002).
Chandrasekharan, N. V. et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc. Natl Acad. Sci. USA 99, 13926–13931 (2002).
Kis, B., Snipes, J. A., Simandle, S. A. & Busija, D. W. Acetaminophen-sensitive prostaglandin production in rat cerebral endothelial cells. Am. J. Physiol. Regulatory, Integr. Comp. Physiol. 288, R897–R902 (2005).
Schwab, J. M., Schluesener, H. J., Meyermann, R. & Serhan, C. N. COX-3 the enzyme and the concept: steps towards highly specialized pathways and precision therapeutics? Prostaglandins, Leukotrienes, Essent. Fat. Acids 69, 339–343 (2003).
Narumiya, S. & FitzGerald, G. A. Genetic and pharmacological analysis of prostanoid receptor function. J. Clin. Investig. 108, 25–30 (2001).
Breyer, R. M., Bagdassarian, C. K., Myers, S. A. & Breyer, M. D. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharm. Toxicol. 41, 661–690 (2001).
O’Callaghan, G. & Houston, A. Prostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br. J. Pharmacol. 172, 5239–5250 (2015).
Coleman, R. A., Smith, W. L. & Narumiya, S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 46, 205–229 (1994).
Jones, R. L., Giembycz, M. A. & Woodward, D. F. Prostanoid receptor antagonists: development strategies and therapeutic applications. Br. J. Pharmacol. 158, 104–145 (2009).
Shimizu, T. et al. Characterization of leukotriene A4 synthase from murine mast cells: evidence for its identity to arachidonate 5-lipoxygenase. Proc. Natl Acad. Sci. USA 83, 4175–4179 (1986).
Lewis, R. A., Austen, K. F. & Soberman, R. J. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N. Engl. J. Med. 323, 645–655 (1990).
Samuelsson, B. The discovery of the leukotrienes. Am. J. Respiratory Crit. Care Med. 161, S2–S6 (2000).
Hanaka, H., Shimizu, T. & Izumi, T. Nuclear-localization-signal-dependent and nuclear-export-signal-dependent mechanisms determine the localization of 5-lipoxygenase. Biochem. J. 361, 505–514 (2002).
Saeki, K. & Yokomizo, T. Identification, signaling, and functions of LTB4 receptors. Semin Immunol. 33, 30–36 (2017).
Funk, C. D. et al. Molecular cloning and amino acid sequence of leukotriene A4 hydrolase. Proc. Natl Acad. Sci. USA 84, 6677–6681 (1987).
Dixon, R. A. et al. Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis. Nature 343, 282–284 (1990).
Matsumoto, T. et al. Molecular cloning and amino acid sequence of human 5-lipoxygenase. Proc. Natl Acad. Sci. USA 85, 26–30 (1988).
Lam, B. K., Penrose, J. F., Freeman, G. J. & Austen, K. F. Expression cloning of a cDNA for human leukotriene C4 synthase, an integral membrane protein conjugating reduced glutathione to leukotriene A4. Proc. Natl Acad. Sci. USA 91, 7663–7667 (1994).
Murphy, R. C. & Gijon, M. A. Biosynthesis and metabolism of leukotrienes. Biochem. J. 405, 379–395 (2007).
Lotzer, K., Funk, C. D. & Habenicht, A. J. The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis. Biochimica et. Biophysica Acta 1736, 30–37 (2005).
Powell, W. S. & Rokach, J. Targeting the OXE receptor as a potential novel therapy for asthma. Biochem. Pharmacol. 179, 113930 (2020).
Back, M., Sultan, A., Ovchinnikova, O. & Hansson, G. K. 5-Lipoxygenase-activating protein: a potential link between innate and adaptive immunity in atherosclerosis and adipose tissue inflammation. Circulation Res. 100, 946–949 (2007).
Snelgrove, R. J. et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science 330, 90–94 (2010).
Wheelan, P. et al. Stereochemical analysis and biological activity of 3-hydroxy-leukotriene B4: a metabolite from ethanol-treated rat hepatocytes. J. Pharmacol. Exp. Ther. 271, 1514–1519 (1994).
Kikuta, Y., Kusunose, E. & Kusunose, M. Characterization of human liver leukotriene B(4) omega-hydroxylase P450 (CYP4F2). J. Biochem. 127, 1047–1052 (2000).
Yokomizo, T. et al. cDNA cloning, expression, and mutagenesis study of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 271, 2844–2850 (1996).
Clish, C. B. et al. Oxidoreductases in lipoxin A4 metabolic inactivation: a novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J. Biol. Chem. 275, 25372–25380 (2000).
Haeggstrom, J. Z. & Funk, C. D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 111, 5866–5898 (2011).
Wang, T. et al. Arachidonic Acid Metabolism and Kidney Inflammation. Int. J. Mol. Sci. 20, 3683 (2019).
Fiore, S., Ryeom, S. W., Weller, P. F. & Serhan, C. N. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J. Biol. Chem. 267, 16168–16176 (1992).
Serhan, C. N. & Sheppard, K. A. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Investig. 85, 772–780 (1990).
Green, A. R. et al. 5 S,15 S-Dihydroperoxyeicosatetraenoic acid (5,15-diHpETE) as a lipoxin intermediate: reactivity and kinetics with human leukocyte 5-lipoxygenase, platelet 12-lipoxygenase, and reticulocyte 15-lipoxygenase-1. Biochemistry 57, 6726–6734 (2018).
Zheng, Z. et al. The biological role of arachidonic acid 12-lipoxygenase (ALOX12) in various human diseases. Biomed. Pharmacother. = Biomedecine pharmacotherapie 129, 110354 (2020).
Dobrian, A. D. et al. Role of the 12-lipoxygenase pathway in diabetes pathogenesis and complications. Pharmacol. Ther. 195, 100–110 (2019).
Singh, N. K. & Rao, G. N. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog. Lipid Res. 73, 28–45 (2019).
Bieche, I. et al. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet. Genomics 17, 731–742 (2007).
Enayetallah, A. E., French, R. A., Thibodeau, M. S. & Grant, D. F. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J. Histochem. Cytochem. 52, 447–454 (2004).
Kiss, L. et al. Direct and simultaneous profiling of epoxyeicosatrienoic acid enantiomers by capillary tandem column chiral-phase liquid chromatography with dual online photodiode array and tandem mass spectrometric detection. Anal. Bioanal. Chem. 392, 717–726 (2008).
Zhang, B., Cao, H. & Rao, G. N. Fibroblast growth factor-2 is a downstream mediator of phosphatidylinositol 3-kinase-Akt signaling in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. J. Biol. Chem. 281, 905–914 (2006).
Wang, Y. et al. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J. Pharmacol. Exp. Ther. 314, 522–532 (2005).
Li, N. et al. Use of metabolomic profiling in the study of arachidonic acid metabolism in cardiovascular disease. Congestive Heart Fail. 17, 42–46 (2011).
Lucotti, S. et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Investig. 129, 1845–1862 (2019).
Marklund, M. et al. Biomarkers of dietary omega-6 fatty acids and incident cardiovascular disease and mortality. Circulation 139, 2422–2436 (2019).
Sonnweber, T. et al. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 19, 3285 (2018).
Vane, J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. N. Biol. 231, 232–235 (1971).
Sanmuganathan, P. S. et al. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 85, 265–271 (2001).
Leal, M. A. S. et al. Sildenafil (Viagra(R)) prevents Cox-1/ TXA2 pathway-mediated vascular hypercontractility in ApoE-/- mice. Cell Physiol. Biochem. 44, 1796–1809 (2017).
Ross, S. J., Elgendy, I. Y. & Bavry, A. A. Cardiovascular safety and bleeding risk associated with nonsteroidal anti-inflammatory medications in patients with cardiovascular disease. Curr. Cardiol. Rep. 19, 8 (2017).
Chan, C. C. et al. Do COX-2 inhibitors raise blood pressure more than nonselective NSAIDs and placebo? An updated meta-analysis. J. Hypertension 27, 2332–2341 (2009).
Ahmad, K. et al. Association of aspirin and other nonsteroidal anti-inflammatory drugs with vertebral trabecular bone: data from multiethnic study of atherosclerosis, a population-based multicenter cohort study. J. Comput. Assist. Tomogr. 44, 562–568 (2020).
MacDonald, T. M. et al. Randomized trial of switching from prescribed non-selective non-steroidal anti-inflammatory drugs to prescribed celecoxib: the Standard care vs. Celecoxib Outcome Trial (SCOT). Eur. Heart J. 38, 1843–1850 (2017).
Nissen, S. E. et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N. Engl. J. Med. 375, 2519–2529 (2016).
Liu, J. Y. et al. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc. Natl Acad. Sci. USA 107, 17017–17022 (2010).
Celotti, F. & Durand, T. The metabolic effects of inhibitors of 5-lipoxygenase and of cyclooxygenase 1 and 2 are an advancement in the efficacy and safety of anti-inflammatory therapy. Prostaglandins Other Lipid Mediators 71, 147–162 (2003).
Fiorucci, S., Meli, R., Bucci, M. & Cirino, G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem. Pharmacol. 62, 1433–1438 (2001).
Alvaro-Gracia, J. M. Licofelone–clinical update on a novel LOX/COX inhibitor for the treatment of osteoarthritis. Rheumatology 43, 21i–25i (2004).
Bias, P., Buchner, A., Klesser, B. & Laufer, S. The gastrointestinal tolerability of the LOX/COX inhibitor, licofelone, is similar to placebo and superior to naproxen therapy in healthy volunteers: results from a randomized, controlled trial. Am. J. Gastroenterol. 99, 611–618 (2004).
Zhao, S., Cheng, C. K., Zhang, C. L. & Huang, Y. Interplay between oxidative stress, cyclooxygenases, and prostanoids in cardiovascular diseases. Antioxid. Redox Signal. (2020). Online ahead of print.
Herrera, M. et al. Complex role for E-prostanoid 4 receptors in hypertension. J. Am. Heart Assoc. 8, e010745 (2019).
Yuhki, K. et al. Roles of prostanoids in the pathogenesis of cardiovascular diseases: novel insights from knockout mouse studies. Pharmacol. Ther. 129, 195–205 (2011).
Jugdutt, B. I. Prostaglandins in myocardial: with emphasis on myocardial preservation. Prostaglandins Med. 7, 109–123 (1981).
Coker, S. J., Parratt, J. R., Ledingham, I. M. & Zeitlin, I. J. Thromboxane and prostacyclin release from ischaemic myocardium in relation to arrhythmias. Nature 291, 323–324 (1981).
Johnson, G. 3rd, Furlan, L. E., Aoki, N. & Lefer, A. M. Endothelium and myocardial protecting actions of taprostene, a stable prostacyclin analogue, after acute myocardial ischemia and reperfusion in cats. Circulation Res. 66, 1362–1370 (1990).
Lefer, A. M. et al. Prostacyclin: a potentially valuable agent for preserving myocardial tissue in acute myocardial ischemia. Science 200, 52–54 (1978).
Schror, K. & Thiemermann, C. Treatment of acute myocardial ischaemia with a selective antagonist of thromboxane receptors (BM 13.177). Br. J. Pharmacol. 87, 631–637 (1986).
Baturova, M. A. et al. Electrocardiographic and Echocardiographic predictors of paroxysmal atrial fibrillation detected after ischemic stroke. BMC Cardiovasc. Disord. 16, 209 (2016).
Calabresi, L. et al. High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-alpha content and enhancing prostaglandin release. Circulation Res. 92, 330–337 (2003).
Berger, H. J. et al. Regional cardiac prostaglandin release during myocardial ischemia in anesthetized dogs. Circulation Res. 38, 566–571 (1976).
Xiao, C. Y. et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation 109, 2462–2468 (2004).
Zacharowski, K. et al. Selective activation of the prostanoid EP(3) receptor reduces myocardial infarct size in rodents. Arterioscler. Thromb. Vasc. Biol. 19, 2141–2147 (1999).
Zacharowski, K., Olbrich, A. & Thiemermann, C. Reduction of myocardial injury by the EP3 receptor agonist TEI-3356. Role of protein kinase C and of K(ATP)-channels. Eur. J. Pharmacol. 367, 33–39 (1999).
Hohlfeld, T., Meyer-Kirchrath, J., Vogel, Y. C. & Schror, K. Reduction of infarct size by selective stimulation of prostaglandin EP(3)receptors in the reperfused ischemic pig heart. J. Mol. Cell Cardiol. 32, 285–296 (2000).
Hara, A. et al. Augmented cardiac hypertrophy in response to pressure overload in mice lacking the prostaglandin I2 receptor. Circulation 112, 84–92 (2005).
Yuhki, K. et al. Roles of prostanoids in the pathogenesis of cardiovascular diseases. Int. Angiol. 29, 19–27 (2010).
Yu, H., Gallagher, A. M., Garfin, P. M. & Printz, M. P. Prostacyclin release by rat cardiac fibroblasts: inhibition of collagen expression. Hypertension 30, 1047–1053 (1997).
Liu, X. et al. Adenylyl cyclase type 6 overexpression selectively enhances beta-adrenergic and prostacyclin receptor-mediated inhibition of cardiac fibroblast function because of colocalization in lipid rafts. Naunyn Schmiedebergs Arch. Pharm. 377, 359–369 (2008).
Deng, X. F., Rokosh, D. G. & Simpson, P. C. Autonomous and growth factor-induced hypertrophy in cultured neonatal mouse cardiac myocytes. Comparison Rat. Circulation Res. 87, 781–788 (2000).
Liu, S. et al. Knockout of the prostaglandin E2 receptor subtype 3 promotes eccentric cardiac hypertrophy and fibrosis in mice. J. Cardiovasc. Pharm. Ther. 22, 71–82 (2017).
Segi, E. et al. Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4-deficient mice. Biochem. Biophys. Res. Commun. 246, 7–12 (1998).
Guan, Y. et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J. Clin. Investig. 117, 2496–2505 (2007).
Kennedy, C. R. et al. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat. Med. 5, 217–220 (1999).
Francois, H. et al. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 43, 364–369 (2004).
Taba, Y. et al. Fluid shear stress induces lipocalin-type prostaglandin D(2) synthase expression in vascular endothelial cells. Circulation Res. 86, 967–973 (2000).
Ho, K. J. et al. Aspirin-triggered lipoxin and resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am. J. Pathol. 177, 2116–2123 (2010).
Mills, C. D. et al. Pillars Article: M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000. 164: 6166-6173. J. Immunol. 199, 2194–2201 (2017).
Merched, A. J. et al. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22, 3595–3606 (2008).
Petri, M. H. et al. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E(-/-) mice. Br. J. Pharmacol. 174, 4043–4054 (2017).
Sekheri, M., El Kebir, D., Edner, N. & Filep, J. G. 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc. Natl Acad. Sci. USA 117, 7971–7980 (2020).
Birnbaum, Y. et al. Augmentation of myocardial production of 15-epi-lipoxin-a4 by pioglitazone and atorvastatin in the rat. Circulation 114, 929–935 (2006).
Campos-Estrada, C. et al. Simvastatin and Benznidazole-Mediated Prevention of Trypanosoma cruzi-Induced Endothelial Activation: Role of 15-epi-lipoxin A4 in the Action of Simvastatin. PLoS Neglected Tropical Dis. 9, e0003770 (2015).
Spite, M. & Serhan, C. N. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circulation Res. 107, 1170–1184 (2010).
Cyrus, T. et al. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Investig. 103, 1597–1604 (1999).
Zhao, L. et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat. Med. 10, 966–973 (2004).
Back, M. & Hansson, G. K. Leukotriene receptors in atherosclerosis. Ann. Med. 38, 493–502 (2006).
Spanbroek, R. et al. Expanding expression of the 5-lipoxygenase pathway within the arterial wall during human atherogenesis. Proc. Natl Acad. Sci. USA 100, 1238–1243 (2003).
Ketelhuth, D. F. et al. The leukotriene B4 receptor (BLT) antagonist BIIL284 decreases atherosclerosis in ApoE-/- mice. Prostaglandins Other Lipid Mediators 121, 105–109 (2015).
Jiang, W. et al. Endothelial cysteinyl leukotriene 2 receptor expression mediates myocardial ischemia-reperfusion injury. Am. J. Pathol. 172, 592–602 (2008).
Hui, Y. et al. Directed vascular expression of human cysteinyl leukotriene 2 receptor modulates endothelial permeability and systemic blood pressure. Circulation 110, 3360–3366 (2004).
Mawhin, M. A. et al. Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxaemia. Cardiovasc. Res. 114, 1656–1666 (2018).
Moos, M. P. & Funk, C. D. Endothelial cysteinyl leukotriene 2 receptor expression and myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 18, 268–273 (2008).
de Hoog, V. C. et al. BLT1 antagonist LSN2792613 reduces infarct size in a mouse model of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 108, 367–376 (2015).
Becher, U. M. et al. Inhibition of leukotriene C4 action reduces oxidative stress and apoptosis in cardiomyocytes and impedes remodeling after myocardial injury. J. Mol. Cell Cardiol. 50, 570–577 (2011).
Ingelsson, E., Yin, L. & Back, M. Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 129, 702–707.e702 (2012).
Hoxha, M., Rovati, G. E. & Cavanillas, A. B. The leukotriene receptor antagonist montelukast and its possible role in the cardiovascular field. Eur. J. Clin. Pharm. 73, 799–809 (2017).
Jamieson, K. L. et al. Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Therapeutics 179, 47–83 (2017).
Luscher, T. F. Cardiomyopathies: definition, diagnosis, causes, and genetics. Eur. Heart J. 37, 1779–1782 (2016).
Sato, M. et al. The roles of cytochrome p450 in ischemic heart disease. Curr. Drug Metab. 12, 526–532 (2011).
Zhao, Q. et al. Endothelium-specific CYP2J2 overexpression improves cardiac dysfunction by promoting angiogenesis via Jagged1/Notch1 signaling. J. Mol. Cell Cardiol. 123, 118–127 (2018).
du Souich, P. & Fradette, C. The effect and clinical consequences of hypoxia on cytochrome P450, membrane carrier proteins activity and expression. Expert Opin. Drug Metab. Toxicol. 7, 1083–1100 (2011).
Keserü, B. et al. Hypoxia-induced pulmonary hypertension: comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc. Res. 85, 232–240 (2010).
Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744 (2018).
Horckmans, M. et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38, 187–197 (2017).
Leuschner, F. et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 209, 123–137 (2012).
Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047 (2007).
Akhnokh, M. K. et al. Inhibition of soluble epoxide hydrolase limits mitochondrial damage and preserves function following ischemic injury. Front. Pharm. 7, 133 (2016).
Aleshin, A. et al. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circulatory Physiol. 294, H1823–H1832 (2008).
Zhang, Y. et al. Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circulatory Physiol. 297, H37–H46 (2009).
Merabet, N. et al. Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J. Mol. Cell Cardiol. 52, 660–666 (2012).
Batchu, S. N. et al. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br. J. Pharmacol. 162, 897–907 (2011).
Chaudhary, K. R. et al. Differential effects of soluble epoxide hydrolase inhibition and CYP2J2 overexpression on postischemic cardiac function in aged mice. Prostaglandins Other Lipid Mediators 104-105, 8–17 (2013).
Gross, G. J. & Nithipatikom, K. Soluble epoxide hydrolase: a new target for cardioprotection. Curr. Opin. Investig. Drugs 10, 253–258 (2009).
Li, R. et al. Cytochrome P450 2J2 is protective against global cerebral ischemia in transgenic mice. Prostaglandins Other Lipid Mediators 99, 68–78 (2012).
Deng, B. Q. et al. Epoxide metabolites of arachidonate and docosahexaenoate function conversely in acute kidney injury involved in GSK3beta signaling. Proc. Natl Acad. Sci. USA 114, 12608–12613 (2017).
Bleske, B. E. et al. Neutral effect on markers of heart failure, inflammation, endothelial activation and function, and vagal tone after high-dose HMG-CoA reductase inhibition in non-diabetic patients with non-ischemic cardiomyopathy and average low-density lipoprotein level. J. Am. Coll. Cardiol. 47, 338–341 (2006).
Oni-Orisan, A., Alsaleh, N., Lee, C. R. & Seubert, J. M. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J. Mol. Cell Cardiol. 74, 199–208 (2014).
He, Z. et al. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc. Res. 105, 304–317 (2015).
Wang, B. et al. CYP2J2 and its metabolites (epoxyeicosatrienoic acids) attenuate cardiac hypertrophy by activating AMPKalpha2 and enhancing nuclear translocation of Akt1. Aging Cell 15, 940–952 (2016).
Nishikimi, T., Maeda, N. & Matsuoka, H. The role of natriuretic peptides in cardioprotection. Cardiovasc. Res. 69, 318–328 (2006).
Xiao, B. et al. Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J. Pharmacol. Exp. Ther. 334, 784–794 (2010).
Yang, L. et al. CYP epoxygenase 2J2 prevents cardiac fibrosis by suppression of transmission of pro-inflammation from cardiomyocytes to macrophages. Prostaglandins Other Lipid Mediators 116-117, 64–75 (2015).
He, Z. et al. CYP2J2 metabolites, epoxyeicosatrienoic acids, attenuate Ang II-induced cardiac fibrotic response by targeting Galpha12/13. J. Lipid Res. 58, 1338–1353 (2017).
Nishida, M. et al. Galpha12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J. Biol. Chem. 282, 23117–23128 (2007).
Tunctan, B. et al. Contribution of iNOS/sGC/PKG pathway, COX-2, CYP4A1, and gp91(phox) to the protective effect of 5,14-HEDGE, a 20-HETE mimetic, against vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock. Nitric Oxide 33, 18–41 (2013).
Althurwi, H. N., Maayah, Z. H., Elshenawy, O. H. & El-Kadi, A. O. Early changes in cytochrome P450s and their associated arachidonic acid metabolites play a crucial role in the initiation of cardiac hypertrophy induced by isoproterenol. Drug Metab. Dispos. 43, 1254–1266 (2015).
Zhao, G. et al. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-alpha-induced injury. J. Lipid Res. 53, 456–466 (2012).
Wang, X. et al. CYP2J2-derived epoxyeicosatrienoic acids suppress endoplasmic reticulum stress in heart failure. Mol. Pharmacol. 85, 105–115 (2014).
Dai, M. et al. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J. Cell Physiol. 230, 2108–2119 (2015).
Ai, D. et al. Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc. Natl Acad. Sci. USA 106, 564–569 (2009).
Xu, D. et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl Acad. Sci. USA 103, 18733–18738 (2006).
Monti, J. et al. Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat. Genet. 40, 529–537 (2008).
Westphal, C. et al. CYP2J2 overexpression protects against arrhythmia susceptibility in cardiac hypertrophy. PLoS ONE 8, e73490 (2013).
Qiu, H. et al. Soluble epoxide hydrolase inhibitors and heart failure. Cardiovasc. Ther. 29, 99–111 (2011).
Imig, J. D. & Hammock, B. D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 8, 794–805 (2009).
Boudina, S. & Abel, E. D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 11, 31–39 (2010).
From, A. M., Scott, C. G. & Chen, H. H. The development of heart failure in patients with diabetes mellitus and pre-clinical diastolic dysfunction a population-based study. J. Am. Coll. Cardiol. 55, 300–305 (2010).
Romashko, M., Schragenheim, J., Abraham, N. G. & McClung, J. A. Epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol. Sci. 37, 945–962 (2016).
Ma, B. et al. Cardiac-specific overexpression of CYP2J2 attenuates diabetic cardiomyopathy in male streptozotocin-induced diabetic mice. Endocrinology 154, 2843–2856 (2013).
Fleming, I. The factor in EDHF: cytochrome P450 derived lipid mediators and vascular signaling. Vasc. Pharm. 86, 31–40 (2016).
Fleming, I. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Trends Cardiovasc. Med. 10, 166–170 (2000).
Bellien, J. et al. Evidence for a basal release of a cytochrome-related endothelium-derived hyperpolarizing factor in the radial artery in humans. Am. J. Physiol. Heart Circulatory Physiol. 290, H1347–H1352 (2006).
Whelton, P. K. et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 138, e426–e483 (2018).
Capdevila, J. H., Falck, J. R. & Imig, J. D. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 72, 683–689 (2007).
Polonikov, A. V. et al. A common polymorphism G-50T in cytochrome P450 2J2 gene is associated with increased risk of essential hypertension in a Russian population. Dis. Markers 24, 119–126 (2008).
Jung, O. et al. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45, 759–765 (2005).
Imig, J. D. et al. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 46, 975–981 (2005).
Capdevila, J. & Wang, W. Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr. Opin. Nephrol. Hypertens. 22, 163–169 (2013).
Zhao, X. et al. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrology 15, 1244–1253 (2004).
Capdevila, J. H. et al. The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J. Biol. Chem. 289, 4377–4386 (2014).
Tiwari, S. et al. Increased renal alpha-ENaC and NCC abundance and elevated blood pressure are independent of hyperaldosteronism in vasopressin escape. Am. J. Physiol. Ren. Physiol. 291, F49–F57 (2006).
Imig, J. D. Epoxyeicosanoids in hypertension. Physiol. Res. 68, 695–704 (2019).
Spiecker, M. et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation 110, 2132–2136 (2004).
Lee, C. R. et al. CYP2J2 and CYP2C8 polymorphisms and coronary heart disease risk: the Atherosclerosis Risk in Communities (ARIC) study. Pharmacogenet. Genomics 17, 349–358 (2007).
Bellien, J. & Joannides, R. Epoxyeicosatrienoic acid pathway in human health and diseases. J. Cardiovasc. Pharm. 61, 188–196 (2013).
Liu, W. et al. CYP2J2 overexpression increases EETs and protects against HFD-induced atherosclerosis in ApoE−/− mice. J. Cardiovasc. Pharm. 67, 491–502 (2016).
Ulu, A. et al. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J. Cardiovasc. Pharm. 52, 314–323 (2008).
Cai, Z. et al. CYP2J2 overexpression increases EETs and protects against angiotensin II-induced abdominal aortic aneurysm in mice. J. Lipid Res. 54, 1448–1456 (2013).
Chen, C. & Wang, D. W. CYP epoxygenase derived EETs: from cardiovascular protection to human cancer therapy. Curr. Top. Med. Chem. 13, 1454–1469 (2013).
Deng, Y. et al. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 25, 703–713 (2011).
Zhang, L. N. et al. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 29, 1265–1270 (2009).
Wang, Y. X., Ulu, A., Zhang, L. N. & Hammock, B. Soluble epoxide hydrolase in atherosclerosis. Curr. Atheroscler. Rep. 12, 174–183 (2010).
Zhang, W. et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J. Cereb. Blood Flow. Metab. 27, 1931–1940 (2007).
Alkayed, N. J. et al. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke 33, 1677–1684 (2002).
Liu, Y. et al. Epoxyeicosanoid signaling provides multi-target protective effects on neurovascular unit in rats after focal ischemia. J. Mol. Neurosci. 58, 254–265 (2015).
Chen, X. et al. Soluble epoxide hydrolase inhibition provides multi-target therapeutic effects in rats after spinal cord injury. Mol. Neurobiol. 53, 1565–1578 (2015).
Zhang, L. et al. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet. Genomics 18, 45–51 (2008).
Fleming, I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediators 82, 60–67 (2007).
Webler, A. C. et al. Cytochrome P450 2C9-induced angiogenesis is dependent on EphB4. Arterioscler. Thromb. Vasc. Biol. 28, 1123–1129 (2008).
Munzenmaier, D. H. & Harder, D. R. Cerebral microvascular endothelial cell tube formation: role of astrocytic epoxyeicosatrienoic acid release. Am. J. Physiol. Heart Circulatory Physiol. 278, H1163–H1167 (2000).
Michael, P. et al. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J. Biol. Chem. 277, 15671–15676 (2002).
Guijun, Y. et al. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc. Res. 78, 308–314 (2008).
Michael, P. et al. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by down-regulating p27Kip1. J. Biol. Chem. 278, 29619–29625 (2003).
Ambra, P. et al. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J. Biol. Chem. 280, 27138–27146 (2005).
Anna, L. S. et al. Cytochrome P450-derived epoxyeicosatrienoic acids accelerate wound epithelialization and neovascularization in the hairless mouse ear wound model. Langenbeck’s Arch. Surg. 396, 1245–1253 (2011).
Megan, E. C. et al. The role of cytochrome P450 epoxygenases in retinal angiogenesis. Investig. Ophthalmol. Vis. Sci. 55, 4253–4260 (2014).
Ausprunk, D. H. & Folkman, J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc. Res. 14, 53–65 (1977).
Michaelis, U. R. et al. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J. Cell Sci. 118, 5489–5498 (2005).
Sun, J. et al. Inhibition of vascular smooth muscle cell migration by cytochrome P450 epoxygenase-derived eicosanoids. Circulation Res. 90, 1020–1027 (2002).
Heissig, B. et al. Angiogenesis: vascular remodeling of the extracellular matrix involves metalloproteinases. Curr. Opin. Hematol. 10, 136–141 (2003).
Teleanu, R. I., Chircov, C., Grumezescu, A. M. & Teleanu, D. M. Tumor angiogenesis and anti-angiogenic strategies for cancer treatment. J. Clin. Med. 9, 84 (2019).
Sommer, K. et al. 11,12 and 14,15 epoxyeicosatrienoic acid rescue deteriorated wound healing in ischemia. PLoS ONE 14, e0209158 (2019).
Chen, J. K., Capdevila, J. & Harris, R. C. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc. Natl Acad. Sci. USA 99, 6029–6034 (2002).
Michaelis, U. R. et al. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J. 17, 770–772 (2003).
Dai, N. et al. Vascular repair and anti-inflammatory effects of soluble epoxide hydrolase inhibitor. Exp. Therapeutic Med. 17, 3580–3588 (2019).
Medhora, M. et al. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am. J. Physiol. Heart Circulatory Physiol. 284, H215–H224 (2003).
Cheranov, S. Y. et al. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 111, 5581–5591 (2008).
Michaelis, U. R. et al. Cytochrome P4502C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler. Thrombosis, Vasc. Biol. 25, 321–326 (2005).
Rand, A. A. et al. Epoxyeicosatrienoic acid (EET)-stimulated angiogenesis is mediated by epoxy hydroxyeicosatrienoic acids (EHETs) formed from COX-2. J. Lipid Res. 60, 1996–2005 (2019).
Spector, A. A. & Norris, A. W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 292, C996–C1012 (2007).
Michaelis, U. R. et al. Role of cytochrome P450 2C epoxygenases in hypoxia-induced cell migration and angiogenesis in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 49, 1242–1247 (2008).
Oguro, A., Sakamoto, K., Suzuki, S. & Imaoka, S. Contribution of hydrolase and phosphatase domains in soluble epoxide hydrolase to vascular endothelial growth factor expression and cell growth. Biol. Pharmaceutical Bull. 32, 1962–1967 (2009).
Guo, Y. et al. TPPU enhanced exercise-induced epoxyeicosatrienoic acid concentrations to exert cardioprotection in mice after myocardial infarction. J. Cell. Mol. Med. 22, 1489–1500 (2018).
Zhao, H. et al. Cytochrome P450 (CYP) epoxygenases as potential targets in the management of impaired diabetic wound healing. Lab. Investig. 97, 782–791 (2017).
Reale, A. et al. Functional and biological role of endothelial precursor cells in tumour progression: a new potential therapeutic target in haematological malignancies. Stem Cells Int. 2016, 7954580 (2016).
Xu, D. Y. et al. A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARγ to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int. J. Cardiol. 167, 1298–1304 (2013).
Deng, L. M. et al. Aerobic exercise-based rehabilitation affects the activities of progenitor endothelial cells through EETs pathway. Med. Hypotheses 85, 1037–1038 (2015).
Wu, C. C. et al. Androgen-sensitive hypertension associates with upregulated vascular CYP4A12-20-HETE synthase. J Am. Soc. Nephrol. 24, 1288–1296 (2013).
Gilani, A. et al. Proximal tubular-targeted overexpression of the Cyp4a12-20-HETE synthase promotes salt-sensitive hypertension in malemice. Am. J. Physiol. Regulatory, Integrative Comparative Physiol. 319, R87–R95 (2020).
Zhang, C. et al. Conflicting roles of 20-HETE in hypertension and renal end organ damage. Eur. J. Pharmacol. 833, 190–200 (2018).
Gebremedhin, D. et al. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J. Physiol. 507(Pt 3), 771–781 (1998).
Fan, F. et al. 20-Hydroxyeicosatetraenoic acid contributes to the inhibition of K+ channel activity and vasoconstrictor response to angiotensin II in rat renal microvessels. PLoS ONE 8, e82482 (2013).
Alonso-Galicia, M. et al. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am. J. Physiol. Regulatory, Integr. Comp. Physiol. 283, R60–R68 (2002).
Sodhi, K. et al. CYP4A2-induced hypertension is 20-hydroxyeicosatetraenoic acid- and angiotensin II-dependent. Hypertension 56, 871–878 (2010).
Garcia, V. et al. 20-HETE activates the transcription of angiotensin-converting enzyme via nuclear factor-kappaB translocation and promoter binding. J. Pharmacol. Exp. Ther. 356, 525–533 (2016).
Zordoky, B. N., Anwar-Mohamed, A., Aboutabl, M. E. & El-Kadi, A. O. Acute doxorubicin cardiotoxicity alters cardiac cytochrome P450 expression and arachidonic acid metabolism in rats. Toxicol. Appl. Pharmacol. 242, 38–46 (2010).
Jarrar, Y. B., Jarrar, Q., Abed, A. & Abu-Shalhoob, M. Effects of nonsteroidal anti-inflammatory drugs on the expression of arachidonic acid-metabolizing Cyp450 genes in mouse hearts, kidneys and livers. Prostaglandins Other Lipid Mediators 141, 14–21 (2019).
Zou, A. P. et al. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K(+)-channel activity. Am. J. Physiol. 270, F822–F832 (1996).
Ye, D. et al. Mechanism of rat mesenteric arterial KATP channel activation by 14,15-epoxyeicosatrienoic acid. Am. J. Physiol. Heart Circulatory Physiol. 290, H1326–H1336 (2006).
Ding, Y. et al. The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the G(s) protein. J. Pharmacol. Exp. Ther. 350, 14–21 (2014).
Ma, S. K. et al. Overexpression of G-protein-coupled receptor 40 enhances the mitogenic response to epoxyeicosatrienoic acids. PLoS ONE 10, e0113130 (2015).
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA 69, 7–34 (2019).
Rothwell, P. M. et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 376, 1741–1750 (2010).
Wang, D. & Dubois, R. N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29, 781–788 (2010).
Dufour, M. et al. PGE2-induced colon cancer growth is mediated by mTORC1. Biochem. Biophys. Res Commun. 451, 587–591 (2014).
McCarty, M. F. Minimizing the cancer-promotional activity of cox-2 as a central strategy in cancer prevention. Med Hypotheses 78, 45–57 (2012).
Yang, P., Jiang, Y. & Fischer, S. M. Prostaglandin E3 metabolism and cancer. Cancer Lett. 348, 1–11 (2014).
Xia, D. et al. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat. Med. 18, 224–226 (2012).
Jiang, J. G. et al. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 65, 4707–4715 (2005).
Jiang, J. G. et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 67, 6665–6674 (2007).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Greten, F. R. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296 (2004).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Wang, X., Baek, S. J. & Eling, T. COX inhibitors directly alter gene expression: role in cancer prevention? Cancer Metastasis Rev. 30, 641–657 (2011).
Prieto, P. et al. Interplay between post-translational cyclooxygenase-2 modifications and the metabolic and proteomic profile in a colorectal cancer cohort. World J. Gastroenterol. 25, 433–446 (2019).
Swami, S. et al. Inhibition of prostaglandin synthesis and actions by genistein in human prostate cancer cells and by soy isoflavones in prostate cancer patients. Int. J. Cancer 124, 2050–2059 (2009).
Karpisheh, V. et al. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediators 144, 106338 (2019).
Hata, A. N. & Breyer, R. M. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Therapeutics 103, 147–166 (2004).
Sonoshita, M. et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat. Med. 7, 1048–1051 (2001).
Fiebich, B. L. et al. Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J. Neurochem. 79, 950–958 (2001).
Mutoh, M. et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 62, 28–32 (2002).
Regan, J. W. EP2 and EP4 prostanoid receptor signaling. Life Sci. 74, 143–153 (2003).
Ricciotti, E. & FitzGerald, G. A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000 (2011).
Buchanan, F. G., Wang, D., Bargiacchi, F. & DuBois, R. N. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J. Biol. Chem. 278, 35451–35457 (2003).
Fujino, H., Xu, W. & Regan, J. W. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 278, 12151–12156 (2003).
Shao, J. et al. Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 63, 5218–5223 (2003).
Han, S. & Roman, J. Suppression of prostaglandin E2 receptor subtype EP2 by PPARgamma ligands inhibits human lung carcinoma cell growth. Biochem. Biophys. Res. Commun. 314, 1093–1099 (2004).
Baron, J. A. Aspirin and NSAIDs for the prevention of colorectal cancer. Recent Results Cancer Res. 181, 223–229 (2009).
Iwama, T. NSAIDs and colorectal cancer prevention. J. Gastroenterol. 44(Suppl 19), 72–76 (2009).
Cha, Y. I. & DuBois, R. N. NSAIDs and cancer prevention: targets downstream of COX-2. Annu. Rev. Med. 58, 239–252 (2007).
Olsen, J. H. et al. Use of NSAIDs, smoking and lung cancer risk. Br. J. Cancer 98, 232–237 (2008).
Zhao, Y. S. et al. Association between NSAIDs use and breast cancer risk: a systematic review and meta-analysis. Breast Cancer Res. Treat. 117, 141–150 (2009).
Piazza, G. A. et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev. Res. (Philos.) 2, 572–580 (2009).
Baek, S. J. et al. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol. Pharmacol. 59, 901–908 (2001).
Zhang, X., Morham, S. G., Langenbach, R. & Young, D. A. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med. 190, 451–459 (1999).
Catalano, A. & Procopio, A. New aspects on the role of lipoxygenases in cancer progression. Histol. Histopathol. 20, 969–975 (2005).
Hong, S. H. et al. Relationship of arachidonic acid metabolizing enzyme expression in epithelial cancer cell lines to the growth effect of selective biochemical inhibitors. Cancer Res. 59, 2223–2228 (1999).
Hennig, R. et al. 5-Lipoxygenase and leukotriene B(4) receptor are expressed in human pancreatic cancers but not in pancreatic ducts in normal tissue. Am. J. Pathol. 161, 421–428 (2002).
Gupta, S. et al. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 91, 737–743 (2001).
Hoque, A. et al. Increased 5-lipoxygenase expression and induction of apoptosis by its inhibitors in esophageal cancer: a potential target for prevention. Carcinogenesis 26, 785–791 (2005).
Li, N. et al. Overexpression of 5-lipoxygenase and cyclooxygenase 2 inhamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clin. Cancer Res. 11, 2089–2096 (2005).
Ye, Y. N. et al. Dual inhibition of 5-LOX and COX-2 suppresses colo cancer formation promoted by cigarette smoke. Carcinogenesis 26, 827–834 (2005).
Narayanan, N. K. et al. Anticancer effects of licofelone (ML-3000) in prostate cancer cells. Anticancer Res. 27, 2393–2402 (2007).
Tavolari, S. et al. Licofelone, a dual COX/5-LOX inhibitor, induces apoptosis in HCA-7 colon cancer cells through the mitochondrial pathway independently from its ability to affect the arachidonic acid cascade. Carcinogenesis 29, 371–380 (2008).
Nixon, J. B. et al. 15-Lipoxygenase-1 has anti-tumorigenic effects in colorectal cancer. Prostaglandins, leukotrienes, Essent. Fat. Acids 70, 7–15 (2004).
Shureiqi, I. et al. 15-LOX-1: a novel molecular target of nonsteroidal anti-inflammatory drug-induced apoptosis in colorectal cancer cells. J. Natl Cancer Inst. 92, 1136–1142 (2000).
Shureiqi, I. et al. Profiling lipoxygenase metabolism in specific steps of colorectal tumorigenesis. Cancer Prev. Res. (Philos.). 3, 829–838 (2010).
Zuo, X. et al. Oxidative metabolism of linoleic acid modulates PPAR-beta/delta suppression of PPAR-gamma activity. Oncogene 25, 1225–1241 (2006).
Kim, J. S. et al. Overexpression of 15-lipoxygenase-1 induces growth arrest through phosphorylation of p53 in human colorectal cancer cells. Mol. Cancer Res. 3, 511–517 (2005).
Shieh, S. Y., Ikeda, M., Taya, Y. & Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 (1997).
Mashima, R. & Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 6, 297–310 (2015).
Sasaki, F. & Yokomizo, T. The leukotriene receptors as therapeutic targets of inflammatory diseases. Int. Immunol. 31, 607–615 (2019).
Chheda, Z. S. et al. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T cell migration into tumors. J. Immunol. 197, 2016–2026 (2016).
Jala, V. R. et al. Leukotriene B4-receptor-1 mediated host response shapes gut microbiota and controls colon tumor progression. Oncoimmunology 6, e1361593 (2017).
Savari, S., Vinnakota, K., Zhang, Y. & Sjolander, A. Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J. Gastroenterol. 20, 968–977 (2014).
Tsai, M. J. et al. Montelukast induces apoptosis-inducing factor-mediated cell death of lung cancer cells. Int. J. Mol. Sci. 18, (2017).
Tsai, M. J. et al. Cysteinyl leukotriene receptor antagonists decrease cancer risk in asthma patients. Sci. Rep. 6, 23979 (2016).
Lukic, A. et al. Exosomes and cells from lung cancer pleural exudates transform LTC4 to LTD4, promoting cell migration and survival via CysLT1. Cancer Lett. 444, 1–8 (2019).
Waxman, D. J., Chen, L., Hecht, J. E. & Jounaidi, Y. Cytochrome P450-based cancer gene therapy: recent advances and future prospects. Drug Metab. Rev. 31, 503–522 (1999).
Spector, A. A., Fang, X., Snyder, G. D. & Weintraub, N. L. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Progress Lipid Res. 43, 55–90 (2004).
Shimizu, S. et al. omega-Hydroxylation of docosahexaenoic acid or arachidonic acid in human colonic well differentiated adenocarcinoma homogenate. Biochimica et Biophysica Acta 1256, 293–296 (1995).
Chen, C. et al. Cytochrome P450 2J2 is highly expressed in hematologic malignant diseases and promotes tumor cell growth. J. Pharmacol. Exp. Ther. 336, 344–355 (2011).
Liang, Z. et al. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea, a selective and potent dual inhibitor of soluble epoxide hydrolase and p38 kinase intervenes in Alzheimer’s signaling in human nerve cells. ACS Chem. Neurosci. 10, 4018–4030 (2019).
Peng, H. et al. Inhibition of soluble epoxide hydrolase in macrophages ameliorates the formation of foam cells- role of heme oxygenase-1. Circulation J. 83, 2555–2566 (2019).
Wang, D. & DuBois, R. N. Measurement of eicosanoids in cancer tissues. Methods Enzymol. 433, 27–50 (2007).
Zhang, Y. et al. An improved ultra-high performance liquid chromatography-tandem mass spectrometry method for simultaneous quantitation of cytochrome P450 metabolites of arachidonic acid in human plasma. J. Chromatogr. A 1563, 144–153 (2018).
Wu, L. et al. PPARα ligand, AVE8134, and cyclooxygenase inhibitor therapy synergistically suppress lung cancer growth and metastasis. BMC Cancer. 19, 1166 (2019).
Skrypnyk, N. et al. PPARalpha activation can help prevent and treat non-small cell lung cancer. Cancer Res. 74, 621–631 (2014).
King, L. M. et al. Cloning of CYP2J2 gene and identification of functional polymorphisms. Mol. Pharmacol. 61, 840–852 (2002).
Xu, M. et al. Cytochrome P450 2J2: distribution, function, regulation, genetic polymorphisms and clinical significance. Drug Metab. Rev. 45, 311–352 (2013).
Lee, S. S. et al. Identification and functional characterization of novel CYP2J2 variants: G312R variant causes loss of enzyme catalytic activity. Pharmacogenet. Genomics. 15, 105–113 (2005).
Marciante, K. D. et al. Common variation in cytochrome P450 epoxygenase genes and the risk of incident nonfatal myocardial infarction and ischemic stroke. Pharmacogenet. Genomics 18, 535–543 (2008).
Börgel, J. et al. The CYP2J2 G-50T polymorphism and myocardial infarction in patients with cardiovascular risk profile. BMC Cardiovasc. Disorders 8, 41 (2008).
Xia, X. L. et al. Research/review: Insights into the mutation-induced dysfunction of arachidonic acid metabolism from modeling of human CYP2J2. Curr. Drug Metab. 15, 502–513 (2014).
Rendic, S. & Di Carlo, F. J. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 29, 413–580 (1997).
Totah, R. A. & Rettie, A. E. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin. Pharmacol. Therapeutics 77, 341–352 (2005).
Jiang, H. et al. Structural and functional insights into polymorphic enzymes of cytochrome P450 2C8. Amino Acids 40, 1195–1204 (2011).
Bahadur, N. et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem. Pharmacol. 64, 1579–1589 (2002).
Soyama, A. et al. Amiodarone N-deethylation by CYP2C8 and its variants, CYP2C8*3 and CYP2C8 P404A. Pharmacol. Toxicol. 91, 174–178 (2002).
van Schaik, R. H. Cancer treatment and pharmacogenetics of cytochrome P450 enzymes. Invest. N. Drugs 23, 513–522 (2005).
Dai, D. et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11, 597–607 (2001).
Hertz, D. L. et al. CYP2C8*3 predicts benefit/risk profile in breast cancer patients receiving neoadjuvant paclitaxel. Breast Cancer Res. Treat. 134, 401–410 (2012).
Barratt, D. T. et al. CYP2C8 genotype significantly alters imatinib metabolism in chronic myeloid leukaemia patients. Clin. Pharmacokinet. 56, 977–985 (2017).
Adehin, A. et al. Inter-individual variation in imatinib disposition: any role for prevalent variants of CYP1A2, CYP2C8, CYP2C9, and CYP3A5 in Nigerian CML patients? Leukemia Lymphoma 60, 216–221 (2019).
Golmohammadzadeh, G., Mohammadpour, A., Ahangar, N. & Shokrzadeh, M. CYP1A1Polymorphisms in Phase I (CYP450) Genes (rs4646421), (rs1056836), (rs749292) and (rs1058930) and Their Relation to Risk of Breast Cancer: A Case-Control Study in Mazandaran Province in North of Iran. Open access Macedonian. J. Med. Sci. 7, 2488–2496 (2019).
Jernström, H. et al. CYP2C8 and CYP2C9 polymorphisms in relation to tumour characteristics and early breast cancer related events among 652 breast cancer patients. Br. J. Cancer 101, 1817–1823 (2009).
Ladero, J. M. et al. Analysis of the functional polymorphism in the cytochrome P450 CYP2C8 gene rs11572080 with regard to colorectal cancer risk. Front. Genet. 3, 278 (2012).
Lee, C. R., Goldstein, J. A. & Pieper, J. A. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 12, 251–263 (2002).
Bozina, N., Bradamante, V. & Lovric, M. Genetic polymorphism of metabolic enzymes P450 (CYP) as a susceptibility factor for drug response, toxicity, and cancer risk. Arhiv za higijenu rada i toksikologiju. 60, 217–242 (2009).
Tranah, G. J. et al. Epoxide hydrolase and CYP2C9 polymorphisms, cigarette smoking, and risk of colorectal carcinoma in the Nurses’ Health Study and the Physicians’ Health Study. Mol. Carcinogenesis 44, 21–30 (2005).
Goldstein, J. A. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br. J. Clin. Pharmacol. 52, 349–355 (2001).
Yao, H. T. et al. The inhibitory effect of polyunsaturated fatty acids on human CYP enzymes. Life Sci. 79, 2432–2440 (2006).
Schwarz, U. I. Clinical relevance of genetic polymorphisms in the human CYP2C9 gene. Eur. J. Clin. Investig. 33, 23–30 (2003).
Kirchheiner, J. & Brockmöller, J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin. Pharmacol. Ther. 77, 1–16 (2005).
Xie, H. G., Prasad, H. C., Kim, R. B. & Stein, C. M. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv. Drug Delivery Rev. 54, 1257–1270 (2002).
London, S. J., Sullivan-Klose, T., Daly, A. K. & Idle, J. R. Lung cancer risk in relation to the CYP2C9 genetic polymorphism among Caucasians in Los Angeles County.Pharmacogenetics 7, 401–404 (1997).
London, S. J. et al. Lung cancer risk in relation to the CYP2C9*1/CYP2C9*2 genetic polymorphism among African-Americans and Caucasians in Los Angeles County, California. Pharmacogenetics 6, 527–533 (1996).
Yasar, U., Eliasson, E. & Dahl, M. L. Association of CYP2C9 genotypes leading to high enzyme activity and colorectal cancer risk. Carcinogenesis 23, 667–668 (2002).
Chan, A. T. et al. A prospective study of genetic polymorphisms in the cytochrome P-450 2C9 enzyme and the risk for distal colorectal adenoma. Clini. Gastroenterol. Hepatol. 2, 704–712 (2004).
Paul, S., Pant, M. C., Parmar, D. & Verma, J. Association and treatment response to capecitabine-based chemoradiotherapy with CYP2C9 polymorphism in head and neck cancer. Ind. J. Cancer 48, 223–229 (2011).
García-Martín, E. et al. Influence of cytochrome P450 CYP2C9 genotypes in lung cancer risk. Cancer Lett. 180, 41–46 (2002).
Liang, S. et al. Meta-analysis of cytochrome P-450 2C9 polymorphism and colorectal cancer risk. PLoS ONE 7, e49134 (2012).
Fortuny, J. et al. Use of analgesics and nonsteroidal anti-inflammatory drugs, genetic predisposition, and bladder cancer risk in Spain. Cancer Epidemiol., Biomarkers Prevention 15, 1696–1702 (2006).
Seredina, T. A. et al. Association of cytochrome P450 genetic polymorphisms with neoadjuvant chemotherapy efficacy in breast cancer patients. BMC Med. Genet. 13, 45 (2012).
Sausville, L. N. et al. The cytochrome P450 slow metabolizers CYP2C9*2 and CYP2C9*3 directly regulate tumorigenesis via reduced epoxyeicosatrienoic acid production. Cancer Res. 78, 4865–4877 (2018).
Roman, R. J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiological Rev. 82, 131–185 (2002).
Guo, A. M. et al. Expression of CYP4A1 in U251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. J. Pharmacol. Exp. Ther. 327, 10–19 (2008).
Guo, M. et al. 9L gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N’-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. J. Pharmacol. Exp. Ther. 317, 97–108 (2006).
Yu, W. et al. Cytochrome P450 omega-hydroxylase promotes angiogenesis and metastasis by upregulation of VEGF and MMP-9 in non-small cell lung cancer. Cancer Chemother. Pharmacol. 68, 619–629 (2011).
Amaral, S. L. et al. CYP4A metabolites of arachidonic acid and VEGF are mediators of skeletal muscle angiogenesis. Am. J. Physiol. Heart Circulatory Physiol. 284, H1528–H1535 (2003).
Yu, W. et al. Increased expression of CYP4Z1 promotes tumor angiogenesis and growth in human breast cancer. Toxicol. Appl. Pharmacol. 264, 73–83 (2012).
Cheng, J. et al. Vascular characterization of mice with endothelial expression of cytochrome P450 4F2. FASEB J. 28, 2915–2931 (2014).
Guo, A. M. et al. The cytochrome P450 4A/F-20-hydroxyeicosatetraenoic acid system: a regulator of endothelial precursor cells derived from human umbilical cord blood. J. Pharmacol. Exp. Ther. 338, 421–429 (2011).
Chen, L. et al. 20-HETE regulates the angiogenic functions of human endothelial progenitor cells and contributes to angiogenesis in vivo. J. Pharmacol. Exp. Ther. 348, 442–451 (2014).
Kobayashi, K., Omori, K. & Murata, T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 37, 347–354 (2018).
Kim, W. et al. Targeting the enzymes involved in arachidonic acid metabolism to improve radiotherapy. Cancer Metastasis Rev. 37, 213–225 (2018).
LaPointe, M. C. & Isenovic, E. Interleukin-1beta regulation of inducible nitric oxide synthase and cyclooxygenase-2 involves the p42/44 and p38 MAPK signaling pathways in cardiac myocytes. Hypertension 33, 276–282 (1999).
Ogata, S. et al. Signaling pathways regulating IL-1alpha-induced COX-2 expression. J. Dent. Res. 86, 186–191 (2007).
Luo, S. F. et al. Involvement of MAPKs, NF-kappaB and p300 co-activator in IL-1beta-induced cytosolic phospholipase A2 expression in canine tracheal smooth muscle cells. Toxicol. Appl. Pharmacol. 232, 396–407 (2008).
Yang, C. M. et al. TNF-alpha induces cytosolic phospholipase A2 expression via Jak2/PDGFR-dependent Elk-1/p300 activation in human lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L543–L551 (2014).
Meliton, A. Y. et al. Cytosolic group IVa phospholipase A2 mediates IL-8/CXCL8-induced transmigration of human polymorphonuclear leukocytes in vitro. J. Inflamm. 7, 14 (2010).
Navarro-Lerida, I., Alvarez-Barrientos, A., Gavilanes, F. & Rodriguez-Crespo, I. Expression of Concern: Distance-dependent cellular palmitoylation of de-novo-designed sequences and their translocation to plasma membrane subdomains. J. Cell Sci. 115, 3119–3130 (2002). J Cell Sci. 130, 3414 (2017).
Wang, D. et al. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 65, 1822–1829 (2005).
Tong, W. G. et al. LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun. 335, 949–956 (2005).
Krysan, K. et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 65, 6275–6281 (2005).
Poligone, B. & Baldwin, A. S. Positive and negative regulation of NF-kappaB by COX-2: roles of different prostaglandins. J. Biol. Chem. 276, 38658–38664 (2001).
Yan, Y., Wang, B., Zuo, Y. G. & Qu, T. Inhibitory effects of mizolastine on ultraviolet B-induced leukotriene B4 production and 5-lipoxygenase expression in normal human dermal fibroblasts in vitro. Photochem. Photobiol. 82, 665–669 (2006).
Fischer, S. M. et al. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol. Carcinog. 25, 231–240 (1999).
Fischer, S. M. et al. Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol. Carcinog. 46, 363–371 (2007).
Black, A. T. et al. UVB light regulates expression of antioxidants and inflammatory mediators in human corneal epithelial cells. Biochem. Pharmacol. 81, 873–880 (2011).
Bachelor, M. A. & Bowden, G. T. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin. Cancer Biol. 14, 131–138 (2004).
Chun, K. S. et al. Curcumin inhibits phorbol ester-induced expression of cyclooxygenase-2 in mouse skin through suppression of extracellular signal-regulated kinase activity and NF-kappaB activation. Carcinogenesis 24, 1515–1524 (2003).
Kundu, J. K., Shin, Y. K., Kim, S. H. & Surh, Y. J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 27, 1465–1474 (2006).
Satpathy, S. R. et al. Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat. Commun. 6, 7064 (2015).
Shen, G. F., Jiang, J. G., Fu, X. N. & Wang, D. W. [Promotive effects of epoxyeicosatrienoic acids (EETs) on proliferation of tumor cells]. Ai zheng = Aizheng = Chin. J. Cancer 27, 1130–1136 (2008).
Chen, C. et al. Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. J. Pharmacol. Exp. Ther. 329, 908–918 (2009).
Colombero, C. et al. Cytochrome 450 metabolites of arachidonic acid (20-HETE, 11,12-EET and 14,15-EET) promote pheochromocytoma cell growthand tumor associated angiogenesis. Biochimie 171-172, 147–157 (2020).
Schmelzle, M. et al. Esophageal cancer proliferation is mediated by cytochrome P450 2C9 (CYP2C9). Prostaglandins Other Lipid Mediators 94, 25–33 (2011).
Mitra, R. et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±)-14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem. 286, 17543–17559 (2011).
Oguro, A., Sakamoto, K., Funae, Y. & Imaoka, S. Overexpression of CYP3A4, but not of CYP2D6, promotes hypoxic response and cell growth of Hep3B cells. Drug Metab. Pharmacokinet. 26, 407–415 (2011).
Katragadda, D. et al. Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J. Mol. Cell. cardiol. 46, 867–875 (2009).
Liu, L. et al. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J. Pharmacol. Exp. Ther. 339, 451–463 (2011).
Allison, S. E. et al. Activation of ALDH1A1 in MDA-MB-468 breast cancer cells that over-express CYP2J2 protects against paclitaxel-dependent cell death mediated by reactive oxygen species. Biochem. Pharmacol. 143, 79–89 (2017).
Guo, Z. et al. Targeting cytochrome P450-dependent cancer cell mitochondria: cancer associated CYPs and where to find them. Cancer Metastasis Rev 37, 409–423 (2018).
Guo, Z. et al. Heme binding biguanides target cytochrome P450-dependent cancer cell mitochondria. Cell Chem. Biol. 24, 1259–1275.e1256 (2017).
Vaidya, K. S. & Welch, D. R. Metastasis suppressors and their roles in breast carcinoma. J. Mammary Gland Biol. Neoplasia 12, 175–190 (2007).
Nithipatikom, K. et al. Inhibition of carcinoma cell motility by epoxyeicosatrienoic acid (EET) antagonists. Cancer Sci. 101, 2629–2636 (2010).
Panigrahy, D. et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 122, 178–191 (2012).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Park, S. A. & Surh, Y. J. Modulation of tumor microenvironment by chemopreventive natural products. Ann. N.Y. Acad. Sci. 1401, 65–74 (2017).
Node, K. et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279 (1999).
Campbell, W. B. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol. Sci. 21, 125–127 (2000).
Tilborghs, S. et al. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Critical Rev. Oncol./Hematol. 120, 141–150 (2017).
Fleming, I. et al. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circulation Res. 88, 44–51 (2001).
Liu, Y. et al. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl Acad. Sci. USA 102, 16747–16752 (2005).
De Palma, M., Biziato, D. & Petrova, T. V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 17, 457–474 (2017).
Frömel, T. et al. Cytochrome P4502S1: a novel monocyte/macrophage fatty acid epoxygenase in human atherosclerotic plaques. Basic Res. Cardiol. 108, 319 (2013).
Chen, X. W. et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 36, 5045–5057 (2017).
Luo, J. et al. 14,15-EET induces the infiltration and tumor-promoting function of neutrophils to trigger the growth of minimal dormant metastases. Oncotarget 7, 43324–43336 (2016).
Schmelzer, K. R. et al. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl Acad. Sci. USA 102, 9772–9777 (2005).
Inceoglu, B. et al. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 79, 2311–2319 (2006).
Schmelzer, K. R. et al. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc. Natl Acad. Sci. USA 103, 13646–13651 (2006).
Mindt, B. C., Fritz, J. H. & Duerr, C. U. Group 2 innate lymphoid cells in pulmonary immunity and tissue homeostasis. Front. Immunol. 9, 840 (2018).
Lambrecht, B. N. & Hammad, H. The immunology of asthma. Nat. Immunol. 16, 45–56 (2015).
Boudaud, M., Turcotte, S., Stankova, J. & Rola-Pleszczynski, M. IL-33 upregulates cysteinyl leukotriene receptor type 1 expression in human peripheral blood CD4(+) T lymphocytes. J. Immunol. 201, 2787–2798 (2018).
Ricciotti, E. et al. Cyclooxygenase-2, asymmetric dimethylarginine, and the cardiovascular hazard from nonsteroidal anti-inflammatory drugs. Circulation 138, 2367–2378 (2018).
Lund, S. J. et al. Leukotriene C4 potentiates IL-33-induced group 2 innate lymphoid cell activation and lung inflammation. J. Immunol. 199, 1096–1104 (2017).
Bankova, L. G. et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc. Natl Acad. Sci. USA 113, 6242–6247 (2016).
Salimi, M. et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J. Allergy Clin. Immunol. 140, 1090–1100.e1011 (2017).
White, A. A. & Stevenson, D. D. Aspirin-exacerbated respiratory disease. N. Engl. J. Med. 379, 1060–1070 (2018).
Smolen, J. S. et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 4, 18001 (2018).
Miyabe, Y., Miyabe, C. & Luster, A. D. LTB4 and BLT1 in inflammatory arthritis. Semin. Immunol. 33, 52–57 (2017).
Bensen, W. et al. Efficacy and safety of valdecoxib in treating the signs and symptoms of rheumatoid arthritis: a randomized, controlled comparison with placebo and naproxen. Rheumatology 41, 1008–1016 (2002).
Abbasi, M. et al. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 234, 10018–10031 (2019).
Chou, R. C. et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33, 266–278 (2010).
Kim, N. D. et al. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J. Exp. Med. 203, 829–835 (2006).
Shao, W. H., Del Prete, A., Bock, C. B. & Haribabu, B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: a critical role for BLT1 in collagen-induced arthritis in mice. J. Immunol. 176, 6254–6261 (2006).
Gyurko, R. et al. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J. Immunol. 177, 7250–7256 (2006).
Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 (2011).
Kanter, J. E. et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl Acad. Sci. USA 109, E715–E724 (2012).
Luo, P. & Wang, M. H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediators 95, 1–10 (2011).
Wang, Y. et al. Prostaglandin F2alpha facilitates hepatic glucose production through CaMKIIgamma/p38/FOXO1 signaling pathway in fasting and obesity. Diabetes 67, 1748–1760 (2018).
Bleich, D. et al. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J. Clin. Investig. 103, 1431–1436 (1999).
McDuffie, M. et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 57, 199–208 (2008).
Imai, Y. et al. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 59, 673–678 (2016).
Zafiriou, M. P. et al. Hepoxilin A(3) protects beta-cells from apoptosis in contrast to its precursor, 12-hydroperoxyeicosatetraenoic acid. Biochimica et. Biophysica Acta 1811, 361–369 (2011).
Ying, W. et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J. Clin. Investig. 127, 1019–1030 (2017).
Tunaru, S. et al. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat. Commun. 9, 177 (2018).
Chen, Y. et al. 20-Iodo-14,15-epoxyeicosa-8(Z)-enoyl-3-azidophenylsulfonamide: photoaffinity labeling of a 14,15-epoxyeicosatrienoic acid receptor. Biochemistry 50, 3840–3848 (2011).
Xu, X. et al. CYP2J3 gene delivery up-regulated adiponectin expression via reduced endoplasmic reticulum stress in adipocytes. Endocrinology 154, 1743–1753 (2013).
Li, R. et al. CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPARgamma. Am. J. Physiol. Endocrinol. Metab. 308, E270–E282 (2015).
Bettaieb, A. et al. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J. Biol. Chem. 288, 14189–14199 (2013).
De Taeye, B. M. et al. Expression and regulation of soluble epoxide hydrolase in adipose tissue. Obesity 18, 489–498 (2010).
Xu, X. et al. Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice. Diabetes 59, 997–1005 (2010).
Luria, A. et al. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc. Natl Acad. Sci. USA 108, 9038–9043 (2011).
Huang, H., Weng, J. & Wang, M. H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediators 125, 80–89 (2016).
Sacerdoti, D., Gatta, A. & McGiff, J. C. Role of cytochrome P450-dependent arachidonic acid metabolites in liver physiology and pathophysiology. Prostaglandins Other Lipid Mediators 72, 51–71 (2003).
Gundala, N. K. V., Naidu, V. G. M. & Das, U. N. Amelioration of streptozotocin-induced type 2 diabetes mellitus in Wistar rats by arachidonic acid. Biochem Biophys. Res. Commun. 496, 105–113 (2018).
Borgeson, E. et al. Lipoxin A4 attenuates adipose inflammation. FASEB J. 26, 4287–4294 (2012).
Das, U. N. Arachidonic acid and lipoxin A4 as possible endogenous anti-diabetic molecules. Prostaglandins, Leukotrienes, Essent. Fat. Acids 88, 201–210 (2013).
Das, U. N. Is there a role for bioactive lipids in the pathobiology of diabetes mellitus? Front. Endocrinol. 8, 182 (2017).
Godson, C. et al. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 164, 1663–1667 (2000).
Martinez, F. O., Sica, A., Mantovani, A. & Locati, M. Macrophage activation and polarization. Front. Biosci. 13, 453–461 (2008).
Cusi, K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr. Diabetes Rep. 10, 306–315 (2010).
Lumeng, C. N., Deyoung, S. M., Bodzin, J. L. & Saltiel, A. R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 56, 16–23 (2007).
Qiu, J., Shi, Z. & Jiang, J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 22, 148–156 (2017).
Marschallinger, J. et al. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 6, 8466 (2015).
Sarau, H. M. et al. Identification, molecular cloning, expression, and characterization of a cysteinyl leukotriene receptor. Mol. Pharmacol. 56, 657–663 (1999).
Ciana, P. et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 25, 4615–4627 (2006).
Burnstock, G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 104, 4–17 (2016).
Brand-Schieber, E., Falck, J. F. & Schwartzman, M. Selective inhibition of arachidonic acid epoxidation in vivo. J. Physiol. Pharmacol. 51, 655–672 (2000).
VanAlstine, M. A. & Hough, L. B. Effects of acetylenic epoxygenase inhibitors on recombinant cytochrome p450s. Drug Metabolism Disposition: Biol. Fate Chemicals 39, 1221–1226 (2011).
Backman, J. T., Filppula, A. M., Niemi, M. & Neuvonen, P. J. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol. Rev. 68, 168–241 (2016).
Doggrell, S. A. Gemfibrozil prevents major coronary events by increasing HDL-cholesterol and more. Expert Opin. Pharmacother. 2, 1187–1189 (2001).
Wiggins, B. S., Saseen, J. J. & Morris, P. B. Gemfibrozil in combination with statins-is it really contraindicated? Curr. Atheroscler. Rep. 18, 18 (2016).
Roy, A. & Pahan, K. Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol. Immunotoxicol. 31, 339–351 (2009).
Shao, J. et al. Involvement of the arachidonic acid cytochrome P450 epoxygenase pathway in the proliferation and invasion of human multiple myeloma cells. PeerJ. 4, e1925 (2016).
Shao, J. et al. P-450-dependent epoxygenase pathway of arachidonic acid is involved in myeloma-induced angiogenesis of endothelial cells. J. Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen ban. 31, 596 (2011).
Floriano-Sanchez, E. et al. CYP3A4 expression in breast cancer and its association with risk factors in Mexican women. Asian Pac. J. Cancer Prevention 15, 3805–3809 (2014).
Sausville, L. N., Williams, S. M. & Pozzi, A. Cytochrome P450 epoxygenases and cancer: A genetic and a molecular perspective. Pharmacol. Ther. 196, 183–194 (2018).
Askari, A. A. et al. Basal and inducible anti-inflammatory epoxygenase activity in endothelial cells. Biochem. Biophys. Res. Commun. 446, 633–637 (2014).
Mittal, B. et al. Cytochrome P450 in cancer susceptibility and treatment. Adv. Clin. Chem. 71, 77–139 (2015).
Murray, M. & Reidy, G. F. Selectivity in the inhibition of mammalian cytochromes P-450 by chemical agents. Pharmacol. Rev. 42, 85–101 (1990).
Sinal, C. J. et al. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 275, 40504–40510 (2000).
Liu, J. Y. et al. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br. J. Pharmacol. 156, 284–296 (2009).
Liu, J. Y. et al. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur. J. Pharm. Sci. 48, 619–627 (2013).
Inceoglu, B. et al. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc. Natl Acad. Sci. USA 105, 18901–18906 (2008).
Harris, T. R., Li, N., Chiamvimonvat, N. & Hammock, B. D. The potential of soluble epoxide hydrolase inhibition in the treatment of cardiac hypertrophy. Congestive Heart Failure 14, 219–224 (2008).
Harris, T. R. et al. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol. Appl. Pharmacol. 286, 102–111 (2015).
Kim, J. et al. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am. J. Physiol. Renal Physiol. 307, F971–F980 (2014).
Meissner, M. et al. PPARalpha activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circulation Res. 94, 324–332 (2004).
Pozzi, A. et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 282, 17685–17695 (2007).
Panigrahy, D. et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl Acad. Sci. USA 105, 985–990 (2008).
Pozzi, A. et al. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J. Biol. Chem. 285, 12840–12850 (2010).
Skrypnyk, N. et al. PPARα activation can help prevent and treat non-small cell lung cancer. Cancer Res. 74, 621–631 (2014).
Denison, M. S. & Whitlock, J. P. Jr. Xenobiotic-inducible transcription of cytochrome P450 genes. J. Biol. Chem. 270, 18175–18178 (1995).
Waxman, D. J. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch. Biochem. Biophys. 369, 11–23 (1999).
Sinz, M., Wallace, G. & Sahi, J. Current industrial practices in assessing CYP450 enzyme induction: preclinical and clinical. AAPS J. 10, 391–400 (2008).
Park, Y. J. et al. Opposing regulation of cytochrome P450 expression by CAR and PXR in hypothyroid mice. Toxicol. Appl. Pharmacol. 263, 131–137 (2012).
Gahrs, M., Roos, R., Andersson, P. L. & Schrenk, D. Role of the nuclear xenobiotic receptors CAR and PXR in induction of cytochromes P450 by non-dioxinlike polychlorinated biphenyls in cultured rat hepatocytes. Toxicol. Appl. Pharmacol. 272, 77–85 (2013).
Smutny, T., Mani, S. & Pavek, P. Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily. Curr. Drug Metab. 14, 1059–1069 (2013).
Akiyama, T. E. & Gonzalez, F. J. Regulation of P450 genes by liver-enriched transcription factors and nuclear receptors. Biochimica et. Biophysica Acta 1619, 223–234 (2003).
Wiwi, C. A. & Waxman, D. J. Role of hepatocyte nuclear factors in growth hormone-regulated, sexually dimorphic expression of liver cytochromes P450. Growth Factors 22, 79–88 (2004).
Jonas, S. & Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433 (2015).
Shahabi, P., Siest, G., Meyer, U. A. & Visvikis-Siest, S. Human cytochrome P450 epoxygenases: variability in expression and role in inflammation-related disorders. Pharmacol. Ther. 144, 134–161 (2014).
Chen, F. et al. Let-7b inhibits human cancer phenotype by targeting cytochrome P450 epoxygenase 2J2. PLoS ONE 7, e39197 (2012).
Yu, D. et al. Suppression of CYP2C9 by microRNA hsa-miR-128-3p in human liver cells and association with hepatocellular carcinoma. Sci. Rep. 5, 8534 (2015).
Rieger, J. K. et al. Inflammation-associated microRNA-130b down-regulates cytochrome P450 activities and directly targets CYP2C9. Drug Metab. Disposition: Biol. Fate Chemicals 43, 884–888 (2015).
Zhang, S. Y. et al. Human CYP2C8 is post-transcriptionally regulated by microRNAs 103 and 107 in human liver. Mol. Pharmacol. 82, 529–540 (2012).
O’Neill, C. P. & Dwyer, R. M. Nanoparticle-based delivery of tumor suppressor microRNA for cancer therapy. Cells 9, 521 (2020).
Willetts, S. & Foley, D. W. True or false? Challenges and recent highlights in the development of aspirin prodrugs. Eur. J. Med. Chem. 192, 112200 (2020).
Mitchell, J. A. et al. Cyclooxygenases and the cardiovascular system. Pharmacol. Ther. 107624, (2020).
Zaman, K. et al. Endogenous S-nitrosoglutathione modifies 5-lipoxygenase expression in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 34, 387–393 (2006).
Masferrer, J. L. et al. Pharmacology of PF-4191834, a novel, selective non-redox 5-lipoxygenase inhibitor effective in inflammation and pain. J. Pharmacol. Exp. Ther. 334, 294–301 (2010).
Orafaie, A., Mousavian, M., Orafai, H. & Sadeghian, H. An overview of lipoxygenase inhibitors with approach of in vivo studies. Prostaglandins Other Lipid Mediators 148, 106411 (2020).
Law, S. W. Y. et al. Neuropsychiatric events associated with leukotriene-modifying agents: a systematic review. Drug Saf. 41, 253–265 (2018).
Di Salvo, E., Patella, V., Casciaro, M. & Gangemi, S. The leukotriene receptor antagonist Montelukast can induce adverse skin reactions in asthmatic patients. Pulm. Pharmacol. Ther. 60, 101875 (2020).
Innes, J. K. & Calder, P. C. The differential effects of eicosapentaenoic acid and docosahexaenoic acid on cardiometabolic risk factors: asystematic review. Int. J. Mol. Sci. 19, 532 (2018).
Sisignano, M., Steinhilber, D., Parnham, M. J. & Geisslinger, G. Exploring CYP2J2: lipid mediators, inhibitors and therapeutic implications. Drug Discov. Today 25, 1744–1753 (2020).
Lee, E. et al. Terfenadone is a strong inhibitor of CYP2J2 present in the human liver and intestinal microsomes. Drug Metab. Pharmacokinet. 33, 159–163 (2018).
Rose, T. E. et al. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem. 53, 7067–7075 (2010).
Chen, D. et al. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 52, 319–328 (2012).
Minamiyama, Y. et al. Vitamin E deficiency accelerates nitrate tolerance via a decrease in cardiac P450 expression and increased oxidative stress. Free Radic. Biol. Med. 40, 808–816 (2006).
Fer, M. et al. Metabolism of eicosapentaenoic and docosahexaenoic acids by recombinant human cytochromes P450. Arch. Biochem. Biophys. 471, 116–125 (2008).
Campos, S. P. et al. Expression of CYP1A1 and CYP1A2 in the liver and kidney of rabbits after prolonged infusion of propofol. Exp. Toxicol. Pathol. 68, 521–531 (2016).
Nagai, K., Fukuno, S., Suzuki, H. & Konishi, H. Higher gene expression of CYP1A2, 2B1 and 2D2 in the brain of female compared with male rats. Pharmazie 71, 334–336 (2016).
Khayat, M. T. & Nayeem, M. A. The role of adenosine A2A receptor, CYP450s, and PPARs in the regulation of vascular tone. Biomed. Res. Int. 2017, 1720920 (2017).
Peng, Y. et al. Andrographolide inhibits breast cancer through suppressing COX-2 expression and angiogenesis via inactivation of p300 signaling and VEGF pathway. J. Exp. Clin. Cancer Res. 37, 248 (2018).
Huang, R. Y. et al. Thromboxane A2 exerts promoting effects on cell proliferation through mediating cyclooxygenase-2 signal in lung adenocarcinoma cells. J. Cancer Res. Clin. Oncol. 140, 375–386 (2014).
Kim, E. Y., Seo, J. M., Cho, K. J. & Kim, J. H. Ras-induced invasion and metastasis are regulated by a leukotriene B4 receptor BLT2-linked pathway. Oncogene 29, 1167–1178 (2010).
Kim, Y. R. et al. Leukotriene B4 induces EMT and vimentin expression in PANC-1 pancreatic cancer cells: involvement of BLT2 via ERK2 activation. Prostaglandins, Leukotrienes, Essent. Fat. Acids 115, 67–76 (2016).
Nguyen, C. H. et al. Cancer cell-derived 12(S)-HETE signals via 12-HETE receptor, RHO, ROCK and MLC2 to induce lymph endothelial barrier breaching. Br. J. Cancer 115, 364–370 (2016).
Ding, X. Z., Tong, W. G. & Adrian, T. E. 12-lipoxygenase metabolite 12(S)-HETE stimulates human pancreatic cancer cell proliferation via protein tyrosine phosphorylation and ERK activation. Int. J. Cancer 94, 630–636 (2001).
Hao, H. et al. Lipoxin A4 suppresses lipopolysaccharide-induced hela cell proliferation and migration via NF-kappaB pathway. Inflammation 38, 400–408 (2015).
Zong, L. et al. Lipoxin A4 attenuates cell invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer. Oxid. Med. Cell Longev. 2016, 6815727 (2016).
Luo, J. et al. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin alphavbeta3 and activating FAK/PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 37, 23 (2018).
Mitra, R. et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±)-14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem. 286, 17543–17559 (2011).
Acknowledgements
B.W. and L.W. designed and wrote the manuscript draft; J.C., L.D., C.C., and Z.W. helped collect references; J.H. and I.F. have edited the revised manuscript and improving the English. D.W.W., corresponding author, provided financial supports, designed the study, and finished the final writing of the manuscript. This work was supported in part by National Nature Science Foundation of China (Nos. 81790624, 81700333, 81900363, and 81900244) and the Deutsche Forschungsgemeinschaft: SFB 1039/A6 (to I.F.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, B., Wu, L., Chen, J. et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Sig Transduct Target Ther 6, 94 (2021). https://doi.org/10.1038/s41392-020-00443-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00443-w
