
Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease

1
College of Pharmacy and Medical Research Center, Chungbuk National University, Cheongju 28160, Korea
2
College of Veterinary Medicine, Jeonbuk National University, Jeonju 54896, Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Antioxidants 2022, 11(1), 91; https://doi.org/10.3390/antiox11010091
Submission received: 30 November 2021
/
Revised: 27 December 2021
/
Accepted: 28 December 2021
/
Published: 30 December 2021
(This article belongs to the Special Issue Oxidative Stress in Hepatic Injury)
Abstract
:Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease worldwide, and scientific studies consistently report that NAFLD development can be accelerated by oxidative stress. Oxidative stress can induce the progression of NAFLD to NASH by stimulating Kupffer cells, hepatic stellate cells, and hepatocytes. Therefore, studies are underway to identify the role of antioxidants in the treatment of NAFLD. In this review, we have summarized the origins of reactive oxygen species (ROS) in cells, the relationship between ROS and NAFLD, and have discussed the use of antioxidants as therapeutic agents for NAFLD.
Keywords:
NAFLD; oxidative stress; ROS; mitochondria; ER stress; peroxisome; Kupffer cells; inflammation; hepatocytes; lipid metabolism; HSC; fibrosis; antioxidants1. Introduction
1.1. Background of NAFLD
Nonalcoholic Fatty Liver Disease (NAFLD) is the most common chronic liver disease, and a large proportion of the population worldwide has it [1]. NAFLD is defined as an accumulation of fat (>5%) in the liver cells in the absence of excessive alcohol consumption or other causes of liver disease, including autoimmune disease, drug-induced conditions, or viral hepatitis [2]. Alcohol-associated Liver Disease (ALD) and NAFLD are the most common causes of chronic liver disease. Although they share a common spectrum of fatty liver/steatosis, steatohepatitis, fibrosis, cirrhosis and hepatocellular carcinoma, several differences exist [3]. Fatty degeneration of hepatocytes occurs more frequently in NAFLD than in ALD. In contrast, inflammatory cell infiltration and venous or intravenous fibrosis and venous sclerosis are more common in ALD than in NAFLD [4]. The spectrum of NAFLD ranges from simple steatosis (Non Alcoholic Fatty Liver or NAFL) to Nonalcoholic Steatohepatitis (NASH). NASH is characterized by inflammation, hepatocyte swelling, and varying degrees of fibrosis and has the potential to progress to cirrhosis and liver cancer (hepatocellular carcinoma (HCC)) [5]. Although the exact cause of NAFLD is not defined, recent studies have shown that oxidative stress (OS) due to insulin resistance and hepatic steatosis may be major causes of NAFLD and may play an important role in the progression to NASH [6]. The first stage of NAFLD/NASH is characterized by liver damage due to insulin resistance, increased fat load on hepatocytes [7]. Fat accumulation in the liver occurs as a result of an imbalance between the rate of influx and the rate of clearance of triglycerides (TGs). Most of the free fatty acids (FFAs) stored as TGs result from increased lipolysis in peripheral tissues. This increased lipolysis is a consequence of hyperinsulinemia and dietary fat that induce insulin resistance (IR), and increased lipogenesis. This can lead to inflammation, OS, lipid peroxidation, and mitochondrial dysfunction in the liver [8]. Therefore, fat accumulation in the liver causes metabolic disorders, leading to excessive mitochondrial ROS production and ER stress, which develops into inflammatory steatohepatitis (NASH). NASH is a pro-inflammatory state that leads to the activation of Kupffer cells (KCs) and astrocytes, which stimulate collagen deposition, leading to liver fibrosis [9,10].
1.2. Role of Oxidative Stress in NAFLD
Reactive oxygen species (ROS) are partially reduced oxygen metabolites and have a strong oxidative capacity [11]. ROS have many physiological activities in intracellular redox signaling and growth regulation [12]. Reactive nitrogen species (RNS) are various nitric oxide-derived metabolites, including the nitroxyl anion, nitrosonium cation, higher oxides of nitrogen, S-nitrosothiols, and dinitrosyl iron complexes [13]. ROS and RNS initiate, mediate and modulate intracellular OS through physiological (prohormonal action) or pathogenic (causing destructive vicious cycles) pathways [12]. The hydroxyl radical (HO¯) is the most reactive among free radicals and contributes significantly to the negative effects of OS. It damages biomolecules, induces lipid peroxidation, and induces breaks in DNA strands [12]. In physiological conditions, the equilibrium between the ROS, RNS, and antioxidants enables cellular crosstalk, control of intracellular functions, cell-to-cell interactions, proliferation, differentiation, migration, and contraction [14]. OS induced by ROS and inflammation are participants in the mechanisms that lead to hepatic cell death and tissue injury [15].
In this review, we summarize the pathophysiology of ROS-induced. We will also review the potential of antioxidants in the treatment of NAFLD.
2. Factors Contributing to Oxidative Stress in Liver
2.1. ER Stress and ROS
The endoplasmic reticulum (ER) is a fine network of tubules that performs many versatile functions in the cell [16]. The ER produces a vast majority of all proteins secreted into the extracellular space, including hormones and cytokines, as well as cell surface receptors and other proteins that interact with the environment [17]. In the ER, the catalytic processes of oxidoreductase Ero1 and NADPH oxidase (NOX) produce ROS.
Reduced glutathione (GSH) is the most abundant thiol protein in eukaryotic cells and acts as a thiol–disulfide redox buffer. The balance of oxidized glutathione (GSSG) and GSH maintain intracellular redox homeostasis. In addition, the GSH/GSSG ratio represents the intracellular redox state index [18]. The oxidative activity of ER oxidoreductin 1 (Ero1) significantly contributes to cellular ROS [11]; its activity is similar to that of a number of ER oxidoreductases, including protein disulfide isomerase (PDI), endoplasmic reticulum protein (Erp)72, and Erp57 [19]. When GSH is oxidized, and the intramolecular level of GSSH is increased, Ero1-α is activated [20]. When Ero1-α is activated, it preferentially oxidizes the domain of protein disulfide isomerase (PDI). The oxidized a domain oxidizes the a’ domain in the molecule to a reduced state, and the reduced a domain of PDI is subsequently oxidized by Ero1-α to a fully oxidized and open form [21]. During oxidative protein folding in the ER, the thiol groups on the cysteines of substrate peptides are oxidized and form disulfide bonds, and hydrogen peroxide (H2O2) is generated as a byproduct (Figure 1). The oxidized a’ domain can be reduced by reduced ERp46 or GSH [19]. The a’ domain of the reduced PDI forms disulfide bonds [22], which contributes significantly to ROS production in cells [18]. A recent study revealed that the Ero1-α expression in the adipose tissue of homocysteinemia (HHcy) mice is upregulated and contribute to the accumulation of H2O2, ER peroxidation and ER stress, thereby exacerbating NAFLD [23].
NADPH oxidase (NOX) is the main enzyme responsible for ROS production. Growing evidence suggests that ROS production by NOX increases during ER stress [24]. The NOX family consists of Nox1–5 and Duox-1 and -2; these proteins promote the oxidation of NADPH to produce superoxide and have biological roles in various tissues [25]. Hepatocytes and hepatic stellate cells (HSCs) express NOX1, NOX2, and NOX4, whereas KCs predominantly express NOX2 [26]. Among the above, NOX4 is the most potent ROS-producing enzyme. NOX4 mainly produces H2O2. Unlike other NOX enzymes, NOX4 has an extended extracytoplasmic loop (E-loop), and because of this loop, the enzyme can be converted from an H2O2 -generating enzyme to O2¯—generating enzyme. A conserved histidine in the third extracytoplasmic loop of NOX4 is responsible for the H2O2-producing activity of the enzyme. Additionally, NOX4 interacts directly with p22 phox, and this interaction is a prerequisite for H2O2 production [27,28]. The conserved C-terminal dehydrogenase domain in NOX4 has constitutive electron transfer activity. In the full-length NOX4 protein, the N-terminal transmembrane portion of the enzyme generates ROS, and this has been shown to facilitate electron transport across the membrane on the opposite side of the membrane [29]. NOX4 has been studied as a ROS-generating factor in NAFLD. For example, a NOX4 inhibitor (GKT137831) was employed to confirm the role of NOX4 in NaAsO₂-induced HSC activation and ROS accumulation. The researchers reported that the expression of p22 phox, a subunit of NOX, was inhibited by GKT137831. Therefore, the inhibition of NOX4 was able to reduce the formation of NOX4-p22 phox dimers. In addition, the expression of collagen-1 and alpha-smooth muscle actin (α-SMA) was reduced by GKT137831. These results demonstrated that the activation of NOX4 can promote ROS generation and lead to HSC activation [30].
In conclusion, ER stress is induced through the misfolding of proteins in the ER, and ROS is generated by NOX during ER stress. The inhibition of ER stress signaling may reduce ROS generation, and this aspect can be targeted for developing new therapies for NAFLD.
2.2. Mitochondria and ROS
Mitochondria are important sources of intracellular ROS [31]. During cellular respiration, mitochondria transfer electrons to oxygen and generate ROS as a byproduct of oxidation [32]. Cellular stress such as hypoxia-reoxygenation and toxic substance processing can lead to excessive mitochondrial ROS (mtROS) production; the generated ROS are rapidly released into the cytoplasm, resulting in cellular damage [32]. mtROS are generated primarily during oxidative phosphorylation of electron transport chains (ETCs) present in the inner mitochondrial membrane [11]. Electrons donated from nicotine adenine dinucleotide (NADH) of ETC complex I (NADH dehydrogenase) and flavin adenine dinucleotide (FADH2) of complex II (succinate dehydrogenase) pass through ETC to complex IV (cytochrome c oxidase), which reduces O2 to water. On the other hand, protons (H⁺) actively migrate from the mitochondrial matrix to the intermembrane space, causing the mitochondrial matrix to increase in negative charge; this also increases the positive charge in the intermembrane space, creating a mitochondrial membrane potential (Δψₘ). This proton motive force causes complex V-ATP synthetase (ATP-ase) to generate ATP from adenosine diphosphate (ADP) and inorganic phosphates as the protons re-enter the mitochondrial matrix via complex V enzymes. During this process, leakage of electrons from complex I (NADH dehydrogenase) and complex III (ubiquinol-cytochrome C reductase) partially reduces oxygen to form peroxides (O2•¯). Subsequently, O2•¯ is rapidly transformed into H2O2 by two dismutases: superoxide dismutase 2 (SOD2) of the mitochondrial matrix and superoxide dismutase 1 (SOD1) in the mitochondrial intermembrane space. Collectively, O2•¯ and H2O2 generated in this process constitute mtROS (Figure 1) [33].
Another cause of mtROS production is an overload of mitochondrial calcium. Although Ca2⁺ is well-known as an important secondary messenger with a role in regulating many cellular physiological functions, Ca2⁺ overload is detrimental to mitochondrial functions and may contribute significantly to mtROS generation. Mitochondrial Ca2⁺ is involved in energy production (ATP), the opening of the mitochondrial permeability transition pore (m PTP), and in inducing and preventing apoptosis.
In NAFLD, mtROS are oxidized to promote the cytoplasmic transport of mtDNA, and the oxidized mtDNA binds directly to NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) to stimulate interleukin (IL)-1β production [34]. ROS production is increased in HFD-fed NASH mice. In these mice, mtDNA is released into the plasma and contributes to the development of NASH by activating the toll-like receptor (TLR) 9. HFD-induced mtDNA release may increase the inflammatory response by activating the cyclic GMP–AMP-Synthase (cGAS)-cyclic GMP–AMP (cGAMP) stimulator of interferon genes (STING) pathway. This suggests that a high-fat diet contributes to the development of NAFLD by inducing OS and triggering the release of mtDNA, thereby accelerating liver damage [35].
Taken together, ROS in mitochondria are generated through ETC and Ca2⁺ overload. When mtROS is increased, a mitochondrial damage-associated molecular pattern (mtDAMP) such as mtDNA is released into and out of the cell due to mitochondrial dysfunction, which can accelerate NAFLD. Therefore, the maintenance of mitochondrial homeostasis through inhibition of mtROS can potentially be applied to treat NAFLD.
2.3. Peroxisome and ROS
Peroxisomes are multipurpose organelles involved in fatty acid α-oxidation, β-oxidation of very-long-chain fatty acids (VLCFA), purine catabolism, and the biosynthesis of glycerolipids and bile acids. Peroxisome-generated ROS accounts for about 35% of total intracellular ROS [36,37]. The main causes of ROS and RNS generation in the peroxisomes are D-amino acid metabolism and peroxisomal β-oxidation [36]. Peroxisomal β-oxidation is an essential regulator of hepatic lipid homeostasis. Acetyl-CoA, derived from peroxisomal fatty acid oxidation, plays an essential signaling role involved in the autophagic degradation of lipids. This suggests that the major source of cytoplasmic acetyl-CoA that maintains hepatic lipid homeostasis is peroxisomal β-oxidation [38]. The first step in the peroxisomal β-oxidation cycle is facilitated by FAD-containing acyl-CoA oxidase, which provides electrons directly to molecular oxygen to generate H2O2. This step begins in the classical peroxisome proliferator-activated receptor (PPAR)α regulatory and inducible β-oxidation helices dealing with straight-chain acyl-CoA, which in all species are catalyzed by a single enzyme, AOX. The disruption of the straight-chain acyl-CoA oxidase gene results in severe microvesicular hepatic steatosis in mice. This results in high levels of fatty acid chains in the serum, hepatomegaly, and steatohepatitis. This can accelerate the development of NAFLD [39]. The second and third steps of peroxisomal β-oxidation are catalyzed by two different dual-function proteins. The second step involves the hydration of enoyl-CoA to 3-hydroxyacyl-CoAs followed by and then dehydrogenation to yield 3-ketoacyl-CoA in the third step. Intracellular stimulation can also result in ROS-related effects on peroxisomes. Peroxisomes respond to a variety of external stimuli, such as fatty acids and pyruvates (such as peroxisomal proliferators); PPARα activation stimulates the proliferation of peroxisomes. Stimulated peroxisomes also show induction of the β-oxidation pathway; however, antioxidant enzymes are not simultaneously induced, which results in an increase in the intracellular H₂O₂ concentration (Figure 1) [40].
D-amino acid oxidase (DAO), which is highly expressed in the kidney and liver, is a flavin adenine dinucleotide (FAD)-dependent peroxisomal enzyme that catalyzes the oxidation of neutral and polar D-amino acids [41]. DAO catalyzes oxidative deamination and simultaneously reduces the cofactor FAD. When DAO is released, amino acids are hydrolyzed to 2-oxo acid (α-keto acid) and ammonia, and the reduced flavin is reoxidized to produce H2O2 [42]. The corresponding 2-oxo acids (α-keto acids), H2O2, and ammonia (the products of the D-amino acid oxidase reaction) are cytotoxic [42,43,44]. It has recently been shown that the D-amino acid oxidase/3-mercaptopyruvate sulfurtransferase (DAAO/3-MST) pathway is involved in the metabolism of hydrogen sulfide (H2S) [45].
In summary, ROS is generated through peroxisomal β-oxidation and DAO. Peroxisomal β-oxidation is an essential factor in the regulation of hepatic lipid homeostasis in NAFLD. Peroxisome-generated ROS may contribute to NAFLD progression.
3. The Mechanism of Oxidative Stress in Hepatic Steatosis
NAFLD progresses from simple steatosis to steatohepatitis, fibrosis, cirrhosis, and then to HCC due to insulin resistance, hepatic OS, and lipotoxicity mediated by a long-term western diet [46]. Simple hepatic steatosis occurs in the initial stages of NAFLD. There is a small amount of fat deposition, and, there, immune cells infiltration and hepatic cell damage are not present. Fat accumulation in more than 5% of hepatocytes leads to impairment in the metabolism system of the liver [47].
A “two-hit” strike theory was put forward by Day and James [48] to provide a theoretical basis for progression from NAFLD to NASH; this theory has been widely accepted. The “first hit” is manifested by simple hepatic steatosis. As the intake of free fatty acid increases triglyceride biosynthesis, it leads to fat accumulation in hepatocytes and a concomitant increase in insulin resistance. Moreover, the export efficiency of free fatty acids, TGs, and cholesterol is reduced severely. Thus, this forms the “first hit” of NAFLD. Oxidative stress is the initiator of the “second hit.” The first blow initiates metabolic derailment in the mitochondria, ER, and peroxisomes in hepatocytes. ROS can inhibit mitochondrial respiratory chain enzymes and inactivate glyceraldehyde-3-phosphate dehydrogenase and membrane sodium channels to induce hepatocellular injury [49]. ROS further aggravates lipid peroxidation, cytokines production and lipid accumulation and promotes inflammation and fibrosis through several protein kinases and nuclear transcription factor activation pathways.
3.1. The Mechanism of ROS-Mediated Oxidative Stress and Lipid Metabolism in Hepatocytes
ROS production is driven by the ETC in mitochondria as part of the energy production process. ROS play a role in redox biology and OS, and these two functions significantly contribute to physiological and pathological conditions. ROS at low concentrations [50] contributes to signaling transmission function [51], cell proliferation and differentiation [52], cell adhesion, and apoptosis. In contrast, high levels of ROS induce pathophysiological conditions. Loss of ROS homeostasis damages cellular lipids, proteins, and DNA since OS is induced by abnormal ROS levels [53]. Therefore, ROS play-acts as an essential role in the maintenance of physiological functions in the cell. Mitochondria are primarily responsible for the production of superoxide anions; thus, mitochondria are important metabolic and signaling hubs that function to maintain liver cell homeostasis, flexibility, and cell survival [54]. The metabolic burden of hepatocytes in a fatty liver is increased by a high intake of free fatty acids, cholesterol, and alcohol in the fatty liver. Dysfunctional electron transport and mitochondrial dynamics impair the lipid metabolism and lead to the downregulation of fatty acid consumption and upregulation of fat accumulation; this causes a further increase in mitochondrial ROS [55] and superoxide production [56]. The above has been confirmed in ALD [57] and NAFLD mouse models [58,59,60]. Thus, the disordered mitochondrial function involved in ROS production is a key factor affecting lipid metabolism.
Hepatocytes are parenchymal cells that constitute 80% of the liver mass [61], and are responsible for substance metabolism, detoxification, and protein production [62,63]. In ALD, NAFLD, and hepatitis C virus infections, ROS-mediated OS directly disrupts the function of mitochondria and ER and impacts fatty acid oxidation, lipid synthesis, and protein synthesis in the hepatocytes [54,64,65,66]. Therefore, OS plays an important role in the pathogenesis of liver disease. Hepatocytes contain 1000–2000 mitochondria to maintain normal metabolic functions [67]. Mitochondria are committed to β-oxidation, tricarboxylic acid cycle [68], ketogenesis, electron chain transfer activity, and ATP synthesis to maintain hepatocyte homeostasis [69]. In healthy hepatocytes, fatty acyl-CoA is transported into mitochondria (carnitine-based transport) and broken down to produce NADH and FADH2, which are further processed by the ETC to facilitate proton efflux, water formation, and ATP production. In NAFLD, fatty acid β-oxidation is induced because of increased fatty acid levels; therefore, ROS, which are byproducts of fatty acid metabolism, accumulate during the progression of β-oxidation and electron transport. Increased ROS in turn trigger insulin sensitivity, lipid metabolism disorders, inflammation, and apoptosis of hepatocytes. High ROS levels impact mitochondrial function, resulting in the blockage of the TCA cycle and fatty acid oxidation in the mitochondria. When the damaged mitochondria are unable to respond to the high levels of free fatty acids (the oxidative metabolism of fatty acids), the synthesis of fat is promoted in the cytoplasm. Insulin resistance promotes OS-driven steatosis. Lipid accumulation and OS collectively promote the development of NAFLD [70]. The maintenance of hepatocyte mitochondrial function is thus crucial for reducing steatosis in NAFLD. In the physiological liver microenvironment, cellular homeostasis is maintained by the clearance of damaged mitochondria by autophagy. Mitophagy is a type of autophagy pathway that helps maintain a constant number of mitochondria and is responsible for removing damaged mitochondria [55,71]. Mitophagy is important for clearing mitochondria damaged by ROS [72,73]. Many studies have focused on the PTEN-induced kinase (PINK)/parkin-dependent mitophagy signaling pathway [74,75]. Under normal conditions, stable PINK1 recruits parkin to mitochondria; parkin has been reported to promote the ubiquitination of mitofusin (Mfn1/2), voltage-dependent anion-selective channel (VDAC), and translocase of the outer membrane (TOM) proteins, and leads to enhanced mitophagy [76]. Low Δψₘ inhibits PINK1 degradation caused by presenilins-associated rhomboid-like (PARL) protein, which also induces mitophagy [76,77]. However, accompanied by excessive ROS production in mitochondria, oxidative stress and cell death are caused, which leads to a higher Δψₘ [78]. This indicates that the ROS-induced increase in Δψₘ is not conducive to the occurrence of mitophagy. A study found that the expression of parkin was decreased in NALFD; in contrast, inhibiting parkin degradation improved mitochondrial function by reducing ROS and malondialdehyde (MDA) levels, increasing antioxidant enzyme activity, and restoring mitophagy flux [79]. Insufficient mitophagy flux indirectly increases the accumulation of lipids driven by ROS in hepatocytes. In an HFD mouse model and an oleic acid (OA)/PA-stimulated cultured cell model, the loss of mitophagy was associated with fat accumulation, increased OS, and inflammation [80]. The fat accumulation, OS, and inflammation are alleviated by mitophagy and depend mainly on the mitochondrial damage mediated by ROS. In short, mitophagy plays a positive role as a protective mechanism against OS damage in hepatic steatosis (Figure 2).
3.2. Regulator of Oxidative Stress and Lipid Metabolism in Hepatocytes
OS occurs when the production of ROS and RNS is very high. Redox reactions of H2O2, O2− or OH− ions with DNA, proteins, and lipids within cells causes oxidative injury [81]. AMP-activated protein kinase (AMPK) is involved in the regulation of a variety of cellular metabolic pathways. AMPK inhibits the activity of lipid metabolism-related enzymes such as acetyl-CoA carboxylase (ACC) and transcription factors such as PPARα, PPARγ, and sterol regulatory element-binding proteins (SREBPs); this alleviates lipid accumulation and promotes fatty acid oxidation [82]. AMPK signaling activity is sensitive to ROS, as both endogenous and exogenous ROS can activate AMPK [83,84]. However, in response to H2O2, ataxia–telangiectasia mutation (ATM) activates the tuberous sclerosis complex 2 (TSC2) via AMPK, which results in the inhibition of mammalian targets of rapamycin (mTOR) and the induction of autophagy. This indicates that ROS activates AMPK but exacerbates cell damage. For instance, Fudan–Yueyang–Ganoderma lucidum (FYGL), extracted from Ganoderma lucidum, increased AMPK and ACC activity and ameliorated an imbalance in oxidation and anti-oxidation to promote a steady state. Thus, AMPK is essential for the maintenance of lipid metabolism in hepatocytes.
H2O2 is a strong oxidizing agent and stimulates lipid accumulation through SREBP1c in vitro [85]. In NAFLD, the upregulation of SREBP1c is associated directly with H2O2-triggered OS. In turn, the overexpression of SREBP1c also enhances ROS levels in hepatocytes, increases lipid synthesis, and activates the NF-kB inflammation pathway [86]. The overexpression of SREBP-1 in prostate cancer cells promoted ROS generation, fatty acid synthase expression, and accumulation [87]. This evidence indicates that ROS promotes hepatic steatosis and is an essential mediator in the development of this condition. The inhibition of ROS is considered an important strategy for the alleviation of lipid accumulation in hepatocytes.
Redox homeostasis mechanisms include oxidant and antioxidant systems, and these systems regulate ROS clearance in many cell types. Antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidases (GPxs) in the cell are primarily responsible for lowering ROS levels [88]. ROS can be neutralized by SODs, which catalyze their conversion to the less active H2O2 molecule, which is further reduced to water by the CAT and GPx enzymes [53,89]. However, enzymes involved in lipogenesis and lipid droplet accumulation were elevated in the cultured SOD1-deficient hepatocytes [90]. A similar mechanism involves carnitine palmitoyltransferase 1 (CPT1); this enzyme is involved in fatty acid metabolism and is positively regulated by SOD1 to prevent lipid accumulation in nasopharyngeal carcinoma [91]. Unlike SOD1, mitochondrial SOD2 is a key component that regulates ROS metabolism in the mitochondrial matrix. The elimination of excessive ROS depends on SOD, and this process is essential to alleviate lipid accumulation and metabolic disorders in damaged hepatocytes.
In NAFLD, abnormal ROS metabolism leads to an imbalance between OS and anti-OS mechanisms in hepatocytes. Nuclear factor erythroid 2-related factor 2 (NRF2) is an antioxidant transcription factor that negatively regulates ROS-mediated OS [92]. Other evidence indicates that NRF2 is also involved in lipid metabolism. Increased NRF2 expression in the nucleus induces antioxidant gene expression and inhibits ROS production induced by FFA; this, in turn, blocks lipogenic transcription factors, such as CCAAT-enhancer-binding proteins (C/EBP)α and PPARγ [93]. However, NRF2(L)-KO mice with liver-specific NRF2 knockout on an HFD showed less liver enlargement, inflammation, and hepatic steatosis compared to the wild-type mice; NRF2 (MHFD macrophage-specific NRF2-knockout) mice on an HFD were comparable to the wild-type mice in this regard and showed no significant differences in these parameters [94]. This is most likely due to the dual role of NRF2. Chambel et al. indicated that activated NRF2 inhibited lipid accumulation in white adipose tissue, limited adipogenesis, induced insulin resistance and glucose intolerance, and increased hepatic steatosis in ob/ob mice [92]. The differences in the ages of the experimental animals may explain the contradictory findings in the studies. In summary, the antioxidant mechanism involving NRF2is important for the elimination of cellular ROS (Figure 2).
3.3. Oxidative Stress-Mediated Hepatocytes Apoptosis
High ROS levels lead to the loss of function of various intracellular organelles such as mitochondria and ER, which can result in the activation of apoptosis and cell death. OS-mediated hepatocyte apoptosis and dysfunction increase the infiltration of KCs and the activation of HSCs during liver damage [95]. ROS cause hepatocyte dysfunction by damaging intracellular macromolecules, including lipids, proteins, and DNA. Additionally, a study based on an in vitro menadione-generated superoxide model reported that ROS could directly activate c-Jun N-terminal kinase (JNK)/c-Jun cell death signaling pathways to induce hepatocyte apoptosis [96]. Importantly, hepatocytes are highly susceptible to mitochondrial injury and cell apoptosis in the presence of diminished antioxidant ability. The overproduction of ROS destroys mitochondrial proteins, phospholipids, and mitochondrial DNA (mtDNA) [97,98]. The depletion of mtDNA in hepatocytes leads to the reduced expression of mitochondrial DNA-encoded polypeptides and thus results in mitochondrial dysfunction [99]. Mitochondrial dysfunction promotes the progression of hepatocyte apoptosis. Apoptosis and necrosis of hepatocytes are obvious features of NASH. Hepatocyte apoptosis was reported to correlate positively with hepatic fibrosis [100]. Research indicated that OS and apoptosis were augmented, and the proliferation of hepatocytes was reduced in high cholesterol-fed mice [101]. It is possible that hepatocytes with loss of organelle function are unable to process lipid droplets and cholesterol adequately, leading to lipid accumulation.
4. The Mechanism of Oxidative Stress of Kupffer Cells in NASH
In simple steatosis, lipotoxicity and fat accumulation cause severe inflammation in the liver, which leads to NASH. Liver macrophages are mainly two types: monocyte-derived macrophages, which are transported into the liver through blood circulation, and liver-specific resident macrophages, known as KCs (Kupffer cells), which are derived from bone marrow progenitor cells (constitute almost 80–90% of all liver macrophages) [60,102]. KCs play an important role in the liver immune response and are also responsible for ROS generation through the activation of NOX2 and TLR signaling [103]. Of note, ROS are key signaling molecules in the progression of the inflammatory response. In NAFLD, the accumulation of fat leads to an increase in the endotoxin/lipopolysaccharide (LPS) levels in the liver, which induces the M1 polarization of KCs. The M1 polarized macrophages produce ROS and inflammatory cytokines. The pro-inflammatory cytokines secreted from macrophages induce infiltration of neutrophils and the activation of HSCs, which aggravates liver damage and then leads to the progression of hepatic fibrosis.
4.1. mtDNA Mediated Kupffer Cells Activation in NASH
OS-mediated hepatocyte apoptosis and dysfunction increase the infiltration of KCs and the activation of HSCs during liver damage [95]. This exacerbates the progression from NAFLD to NASH and HCC. Steatosis promoted by OS is the main cause of liver cell apoptosis. ROS prevent the replication and transcription of mtDNA, which leads to mitochondrial dysfunction, and further causes increased ROS production and further mtDNA injury [104]. mtDNA is released from the liver cells and is recognized by the innate immune cells (including resident KCs and monocyte-derived macrophages), leading to an inflammatory response; this further promotes the progression of NASH.
In general, NAFLD ranges from simple steatosis to NASH, and the pathogenesis of NASH is still unclear. Recent research has revealed high serum levels of mtDNA and mitochondria in mouse models and NASH patients. KCs activated by mtDNA secrete pro-inflammatory factors (tumor necrosis factor (TNF)-α and IL-6) through TLR9 and stimulate IFN genes (STING) [105,106,107]. STING is a universal receptor for cyclic dinucleotides, including the bacterial second messengers 3′-5′-cyclic-di-adenosine-monophosphate (CDA), 3′-5′-cyclic-di-guanosine-monophosphate (CDG), and the newly discovered metazoan second messenger ring GMP–AMP [108]. The STING pathway is activated by mtDNA and operates in the liver as well as in other tissues, such as in the lungs (in sepsis-induced acute lung injury) and kidneys (in acute kidney injury) [109,110]. In the NASH mouse model, the lack of STING alleviates hepatic steatosis, fibrosis, and nuclear factor (NF)-κB-dependent inflammation even in the presence of mtDNA-induced stimulation of KCs. The mtDNA derived from hepatocytes is a vital factor that drives the innate immune responses of KCs and the activation of HSCs to accelerate NASH and hepatic fibrosis (Figure 3). As mentioned above, the release of mtDNA constitutes a DAMP and is a response to mitochondrial dysfunction caused by ROS-mediated OS in hepatocytes. A previous study found that KCs also reduced the Δψₘ under the stimulation of saturated fatty acids (palmitic acid) and increased the release of mtDNA from the mitochondria to the cytoplasm, leading to an increase in the expression of NLRP3 inflammasome and the secretion of IL-1β [111]. The burst of OS induces the release of mtDNA from hepatocytes or KCs itself; this event is important for KC activation through the STING pathway.
4.2. Oxidative Stress Mediated Inflammation in Kupffer Cells
In ALD and NAFLD, intestinal endotoxin/LPS is the main stimulator of KC activation, which results in the production of pro-inflammatory factors, chemokines and abundant ROS via TLR4 receptor signaling [65,112]. Although hepatocytes and KCs are the main sources of ROS production, hepatocytes have a higher antioxidant capacity than KCs [113]. It is possible that KCs may have a greater impact on the ROS-mediated mechanisms compared to hepatocytes. The reason may be the high expression of uncoupling protein 2 (UCP2) in hepatocytes and the reduction in KCs in HFD-fed mice [114,115]. UCP2, a mitochondrial inner membrane protein, reduces the Δψₘ and prevents OS-mediated damage by increasing proton flux and ATP synthesis [116]. Research confirms that when Δψm is dissipated by chemical uncoupling agents or overexpression of mitochondrial uncoupling proteins, ROS production is reduced [33,117]. LPS treatment of macrophages with a targeted disruption of UCP2 led to ROS over-production and pro-inflammatory cytokine secretion through NF-kB pathway activation [118]. Therefore, UCP2 hinders the activation of KCs, and this depends on the negative regulation of Δψₘ and ROS levels. Apart from ROS generated by dysfunctional mitochondria, peroxisomes and the microsomal cytochrome P450 system also generate ROX, which affect redox metabolism in KCs.
When KCs and other macrophages in the liver respond to LPS or other stimuli, they rapidly release ROS through the specialized phagocyte oxidase gp91phox or NOX2 systems [119]. The activated NOX2 produces superoxide, which sends signals to thioredoxin, protein kinase C, extracellular signal-regulated kinase (ERK) family members, and NF-κB to respond to the stimulus [120]. Thus, ROS increase the production of pro-inflammatory factors such as TNF-α, IL-6, and IL-1β. NOX2 activation involves a series of protein–protein interactions; phosphorylation of p47phox allows it to interact with p22phox that is constitutively bound to NOX2. p47phox then recruits additional proteins such as p67phox, p40phox, and Rac GTPases to form a complex with NOX2, which leads to NOX2 activation [121]. This protein complex with activated NOX2 generates superoxide ions by transferring an electron from NAPDH to oxygen. The gene ablation of NOX2 leads to a decrease in ROS production, and attenuates M1 activation and promotes M2 polarization of macrophages in the liver.
FFAs and cholesterol contribute to the formation of fatty “foamy” KCs, and can further lead to NASH [122]. FFAs directly promote the M1 polarization of macrophages that results in a pro-inflammatory phenotype [123,124,125]. Excessive FFAs not only aggravate lipid accumulation in hepatocytes but also damage β-oxidation and mitochondrial function in KCs [125]. Studies showed that KCs sense FFAs and promote lipid accumulation in hepatocytes via TNF-α secretion [126]. It is common knowledge that foam cell formation is the main event in the pathogenesis of atherosclerosis [127]. Interestingly, ROS also promote the formation of foam cells [128], and foam cells were also found in the liver of a NAFLD mouse model [129]. Foam cells in the liver may arise from liver-resident KCs, infiltrating macrophages, or a combination of both the cell types [130]. However, whether ROS participates in the formation and regulation of foam cells and during NAFLD remains unclear.
In the antioxidant system, antioxidant proteins and phase II detoxification enzymes (heme oxygenase-1 (HO-1) and NADPH-quinone oxidoreductase-1 (NQO1)) are key protective agents against oxidative damage [131]. Studies have found that the upregulation of HO-1 induced by ethanol involves the activation of JNK-1, hypoxia-inducible factor (HIF)-1α and NRF2, but the expression of NQO1 is only regulated by NRF2 [129]. By upregulating antioxidant genes, NRF2can also protect the liver from inflammation in a ROS-dependent manner [132]. Although NRF2 is elevated under external stimulation, it is downregulated in NASH patients [133]. A recent study determined that inflammation and metabolic deterioration were aggravated in a myeloid-specific NRF2-deficient NASH mouse model [134]. In another study, LPS accelerated the inflammatory response in P62 and NRF2-double knockout KCs [135]. The antioxidant system is indispensable for the inflammatory responses of KCs (Figure 3). This system balances the redox state and alleviates the inflammatory responses mediated by ROS.
5. The Mechanism of Oxidative Stress in Hepatic Fibrosis
The accumulation of fat within the liver triggers chronic inflammation mediated by activated KCs and other infiltrating immune cells; this leads to hepatic fibrosis. About 9% to 25% of NASH patients will develop cirrhosis in 10 to 20 years [136]. Hepatic fibrosis is characterized by the activation of HSCs; this leads to the proliferation and migration of HSCs and results in a fibrotic phenotype (myofibroblasts) [137]. The activated HSCs further promote the formation of excess collagen and the accumulation of extracellular matrix (ECM). TLR activity [138], autophagy [139], ER stress [140], OS [141], and extracellular signals from hepatocytes and KCs and other molecular signaling pathways have been reported to be involved in the activation of HSCs [142,143]. In this section, we focus primarily on the mechanism of HSC activation mediated by OS.
5.1. ROS-Mediated Fibrogenesis in Stellate Cells
OS results from the alteration of the oxidation and antioxidant balance and involves changes in ROS and RNS levels. ROS not only increase hepatocyte metabolism disorders and KC activation but also stimulates the production of collagen I, which is a key intracellular signaling mediator in the TGF-β-driven fibrosis [144]. TGF-β is an important cytokine that induces the activation of HSCs. Under normal conditions, TGF-β participates in wound healing and angiogenesis. However, excess TGF-β is released from damaged hepatocytes and pro-inflammatory KCs [145,146]. This released TGF-β is recognized by the TGF-βRI receptor on the HSCs and increases fibrogenesis via the SMAD2/3 pathway [141]. In this process, phosphorylated SMAD 2/3 also activates the expression of NOX4 [147,148]. Interestingly, NOX enzyme complexes and the mitochondria are the main producers of endogenous ROS [149]. Moreover, studies have found that the expression of NOX4 is upregulated in many fibrosis models [150]. NOX4 knockdown reduces the activation of HSCs without affecting the expression of the TGF-β1 receptor and the phosphorylation of SMAD2, which indicates that NOX4-mediated ROS occur downstream of the TGF-β1 and SMAD complex [151]. The downregulation of HSC activation depends largely on the lack of NOX4 (which indicates a lack of excessive ROS). In addition to NOX4, NOX1 and NOX2 are also expressed in HSCs. The depletion of NOX1 or NOX2 can reduce inflammation and fibrosis caused by carbon tetrachloride (CCl4) and bile duct ligation (BDL) [152,153]. In contrast, NOX4 does not need to recruit cytoplasmic structural subunits to form active enzymes to generate ROS [64]. This implies that NOX4 produces ROS more rapidly than NOX1 and NOX2. Surprisingly, H2O2 and other ROS can directly induce collagen α1 activity [145,154]. Therefore, OS due to ROS plays a key role in TGF-β-mediated fibrogenesis in HSCs (Figure 4).
A study has found that the components of the NLRP3 inflammasome are expressed in HSCs and contribute to the activation of HSCs [155]. NOX4-mediated ROS production activates the NLRP3 inflammasome and hinders the antioxidant system, leading to HSC activation and fibrosis [156]. TGF-β-induced ROS produced by mitochondria can stimulate NLRP3 activation to increase the levels of α-SMA, connective tissue growth factor (CTGF), and tissue inhibitor matrix metalloproteinase 1 (TIMP1), whereas knockout of p66Shc, an oxidoreductase, eliminated this effect [157]. A recent study indicated that the knockout of NLRP3 downregulated ROS production and the mRNA and protein levels of fibrosis markers in HSCs [158]. These data indicate that ROS increases the activation of fibrosis through the activated NLRP3 inflammasome. However, these data do not rule out the role of secreted mature IL-1β and IL-18 in fibrosis.
5.2. The Role of Antioxidants System in Stellate Cells
Hepatic fibrosis is characterized by chronic liver damage caused by excessive deposition of ECM-collagen initiated by HSCs. Platelet-derived growth factors (PDGF) and TGF-β1 are key mediators that trigger the activation of HSCs. Moreover, OS is involved in the cytokine-mediated activation of HSCs and in the process of liver fibrosis. Certainly, the antioxidant system acts to remove intracellular ROS and maintains the normal physiological activity of HSCs. In many cell types, NRF2, which is the most important antioxidant transcription factor in the cell, transcriptionally modulates more than 500 genes, most of which are involved in cell protection mechanisms [159]. These antioxidant genes include SOD, CAT, GPx, GSH, HO-1, and NQO-1 [160]. Oxidative or electrophilic stress causes the dissociation of Keap1 from NRF2, followed by the nuclear translocation of NRF2; NRF2 subsequently modulates gene expression in the nucleus [161]. These genes are transcribed to block the activation of HSCs mediated by ROS such as H2O2, O2− and OH-; this alleviates the accumulation of ECM [160]. During the development of NASH, the NRF2-mediated antioxidant system is unable to adequately limit the ROS overload stimulated by PDGF and TGF-β1 (secreted by KCs and HSCs). Antioxidant targeted- NRF2 increases the expression of heme oxygenase 1 (HMOX1), NQO1, and glutathione S-transferase mu3 (GSTM3) genes, and downregulates NOX1, NOX4, and α-SMA in activated HSC models [162,163]. In NRF2-deficient HSCs, the expression of α-SMA and ECM increases, which involves the upregulation of the TGF-β1/ SMAD pathway [159]. Thus, NRF2 is a potential therapeutic target to prevent the activation of HSCs by ROS (Figure 4).
6. Antioxidants as Treatment for NAFLD
6.1. Flavanols (flavan-3-ols)
Flavonoids are divided into several subtypes according to the substitution pattern on the C ring, which depends on the degree of oxidation of the carbon to which the ring is attached [164]. Flavonoids are mainly the following types: flavones, flavonols, flavanones, flavanonols, isoflavones, and flavan-3-ols [165]. These compounds are known to have hepatoprotective effects based on their anti-inflammatory, antioxidant, blood-sugar-lowering, and hepatic lesion suppression activities. They also function as scavengers of numerous active substances such as peroxide and hydrogen peroxide; CYP2E1 is one of the molecules responsible for the production of these active substances [166]. Flavanols such as catechins have been shown to exert several beneficial effects in NAFLD. Epigallocatechin-3-gallate (EGCG) is a catechin found in green tea [167]. The effect of EGCG intake on GSH levels, lipid peroxidation, and the expression of CYP2E1 was studied in an HFD mouse model. The results demonstrated that EGCG improved fat accumulation and inflammation in the HFD mice [168]. The effect of EGCG in inhibiting HSC activation has been demonstrated in several in vitro studies. A recent study showed that intraperitoneally administered EGCG could reduce liver fibrosis in a rodent model of NASH by inhibiting NF-kB, Akt, and TGF/SMAD signaling as well as OS [169]. EGCG significantly decreased the levels of ROS and MDA in rat serum and liver tissue and increased SOD (Mn SOD, Cu/Zn SOD) enzyme activity and the mRNA levels of GSH-Px. In addition, EGCG decreased the levels of inflammatory factors and the mRNA expression of TNF-α and IL-6 in serum and liver tissue [170].
6.2. Curcumin
Curcumin is a natural polyphenol with antioxidant and anti-inflammatory properties [171]. The antioxidant properties of curcumin have been extensively studied. This compound has been reported to have positive effects on NAFLD and liver metabolism [172]. In a recent study, curcumin effectively reversed the expression of CYP3A and CYP7A in the fatty liver, restoring metabolic capacity [173]. In addition, curcumin exerts a protective effect against inflammatory stimuli by downregulating the expression of inflammation-related transcription factors and reducing the activity of related signaling pathways against linoleic acid and leptin-induced stimuli. Curcumin has a prophylactic effect by targeting circulating monocytes, hepatic macrophages, and peripheral and hepatic CD4+ cells. Curcumin administration inhibited NAFLD development in a mice model of HFD-induced obesity and glucose intolerance and ameliorated histological changes, including fibrosis and intrahepatic accumulation of CD4+ cells [174]. Curcumin exerts beneficial effects on fatty liver disease by reducing hepatic lipid accumulation through the modulation of the AMPK/ACC pathway. O-GlcNAcylation was increased in mice fed the methionine and choline diet (MCD); the upstream target inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) was induced by ER stress, and this factor further upregulated N-acetylglucosaminyltransferase (OGT) and glutamine: fructose-6-phosphate amidotransferase (GFAT).
These results suggest that the induction of OGT and GFAT by the transcriptional regulation of X-box binding protein 1 (XBP1) (which is as an upstream activator of hexosamine biosynthetic pathway (HBP)) activates a positive regulatory loop in NASH pathogenesis. Curcumin effectively inhibited O-GlcNAc protein modification and further downregulated ER stress by inhibiting XBP-related IRE1α and GFAT expression [175].
6.3. Vitamin E
Vitamin E is a lipid-soluble chain-breaking antioxidant (breaks the chain of lipid oxidation) that prevents the production of free radicals. Vitamin E can only be synthesized by plants and exists in the form of four fat-soluble tocopherols and eight forms of tocotrienols that exhibit antioxidant activity [176]. This vitamin has unique antioxidant activity, and the chromanol ring in its structure provides hydrogen ions to free radicals, which results in lipid peroxyl radical scavenging activity. It also increases the activity of other antioxidant enzymes such as catalase and glutathione peroxidase [177]. α-Tocopherol is the most abundant form of vitamin E in nature and the most common and dominant form in human tissues and plasma [178]. In obese (ob/ob) mice, α- or γ-tocopherol showed a protective effect against LPS-induced liver damage and decreased the expression of liver MDA and TNF-α [179]. In addition, vitamin E improved liver necrosis when CCl4 was administered to rats. Vitamin E downregulated the expression of TGF-β1, which is a cytokine implicated in the pathogenesis of liver fibrosis [180].
6.4. Metformin
Metformin (N,N-dimethylbiguanide) belongs to the biguanide class of antidiabetic drugs. Several studies have reported that metformin treatment slows the progression of NAFLD [181]. Metformin has been reported to protect HepG2 cells and primary murine hepatocytes from palmitate- and stearate-induced apoptosis. Metformin partially inhibited mitochondrial respiration, restored Δψₘ, decreased ROS production, and induced SOD2 expression. Metformin partially inhibits mitochondrial complex I, thus preventing mitochondrial dysfunction; it also protects the cell from palmitate-induced cell death [182]. In addition, four weeks of metformin treatment (50 or 100 mg/kg) in obese mice also reduced hepatocyte apolipoprotein A5 (ApoA5) expression; this protein plays a key role in the regulation of TG metabolism and lipid droplet formation [6]. In another study, metformin (200 mg/kg) significantly reduced plasma TG levels, decreased hepatic very-low-density lipoprotein (VLDL)-TG production, and lowered hepatic lipid composition in the APOE*3-Leiden CETP mouse model [183]. Interestingly, metformin (250 mg/kg) enhanced AMPK activation. Metformin-catalyzed increased AMPK phosphorylation suppressed SREBP-1c expression, thereby regulating lipid and glucose metabolism [184]. AMPK also activated NRF2 by directly phosphorylating it or by inhibiting glycogen synthase kinase (GSK)3β, an inhibitory regulator of NRF2 [185].
6.5. Coenzyme Q10
Coenzyme Q10 (CoQ10) is a fat-soluble active quinone that contains a benzoquinone ring with an isoprenoid side chain. CoQ10 is distributed in organelles such as the Golgi apparatus, ER, and lysosomes; 40–50% of the total cellular content of coenzyme Q10 is localized in the inner mitochondrial membrane [186]. The most important feature of CoQ10 is the strong antioxidant capacity of its redox forms (ubiquinone, semi-ubiquinone, ubiquinol) that co-exist in the mitochondrial membrane. CoQ10 exhibits anti-lipogenic properties and thus may have a positive effect on NAFLD. It has been proposed that CoQ10 may regulate hepatic lipid metabolism by inducing AMPK activation (inhibition of adipogenesis and activation of fatty acid oxidation), thereby inhibiting excessive accumulation of hepatic lipids and preventing NAFLD progression [187]. In addition, CoQ10 supplementation was shown to improve liver biochemical enzymes in NASH-induced mice. CoQ10 may modulate TNF-α and adiponectin levels to ameliorate inflammation and improve lipid homeostasis in the liver [188]. For example, in young swimmers, intake of CoQ10 (300 mg/day for 12 days) reduced oxidative stress and the plasma levels of inflammatory markers such as TNF-α and IL-6, and increased the activity of antioxidant enzymes. Similarly, dietary CoQ10 (during 26 weeks of diet supplemented with 0.07–0.7% (w/w) CoQ10) induced the reduction of plasma OS and inflammatory markers in a murine model of the metabolic syndrome [189].
The mechanisms and roles of antioxidants in the treatment of NAFLD have been summarized in Table 1.
6.6. Diet
Insulin resistance associated with diets, especially high-fat or high-carbohydrate diets, increase oxidative stress and the risk of NAFLD [202]. Saturated fatty-acids-enriched foods, not unsaturated fatty acids, such as processed meat or high-fat dairy, induce fat accumulation and insulin resistance, which promotes NAFLD progression [203]. NAFLD patients prefer to follow Western eating habits. Obviously, improving the diet style may be the fundamental way to prevent NAFLD to NASH and hepatic cirrhosis through the metabolic homeostasis connected by the liver–gut axis.
The intake of unsaturated fatty acids instead of saturated fatty acids and isocaloric foods with lower fat content can reduce effectively fat deposition in the liver. In addition, the European Society for Clinical Nutrition and Metabolism (ESPEN) and American Gastroenterology Association (AGA) propose the Mediterranean-style diet [204,205]. The Mediterranean diet is evaluated as a diet style that is beneficial to NAFLD. It did not increase intrahepatic lipid content and transaminase levels, although it is iso-caloric or there are no changes in body weight [206,207]. Remarkably, The Mediterranean diet is characterized by rich antioxidants and fiber-balanced lipid and low sugar content [208]. As we have shown above, the antioxidants in the Mediterranean diet effectively protect the liver from oxidative stress, which has a negative effect on the development of NAFLD. Apart from healthy diets, reasonable exercise seems indispensable. An individual lifestyle with a healthy diet and increased exercise are general interventions in reversing NAFLD progression [204,205]. Shortly, a reasonable diet, including rich fruits, vegetables, and protein, is effective against fat-mediated oxidative damage.
7. Conclusions
NAFLD is characterized by the accumulation of excess fat in the liver, which leads to liver damage. Fat accumulation in NAFLD is not caused by alcohol consumption. Hepatocytes are the main parenchymal cells in the liver, accounting for about 80% of the liver mass. Non-parenchymal cells include resident KCs, HSCs, liver sinusoidal endothelial cells, and other immune cells. These cells contribute to steatosis, inflammation, and fibrosis and promote the progression of NAFLD to NASH and HCC.
Redox reactions are essential for cell survival and for the maintenance of normal liver function. Due to excess fat intake in NAFLD, there is a significant derailment of redox reactions. Energy metabolism occurs primarily in mitochondria; abnormal redox reactions lead to energy metabolism disorders and subsequently increase mitochondrial dysfunction. ROS are increased during mitochondrial oxidative damage and are formed as byproducts of redox reactions. The production of ROS is not limited to mitochondria, and the ER and peroxisome also generate ROS. The upregulation of β-oxidation reactions due to fatty acid overload increases the production of ROS. However, the key mechanism of NAFLD involves OS-induced damage to the hepatocytes, KCs, and HSCs.
Hepatocytes, KCs, and HSCs contribute to the development of NAFLD. ROS promotes lipid accumulation in hepatocytes through a variety of signaling pathways. The defense mechanisms in hepatocytes (including the antioxidant system and mitophagy) are unable to clear ROS and the damaged mitochondria in a timely manner. mtDNA released by hepatocytes subjected to oxidative damage activates pro-inflammatory KCs through the cGAS/STING pathway. Additionally, inflammatory responses in KCs are also induced by intracellular ROS. This is an important mechanism for the progression of NAFLD to NASH. TGF-β released by damaged hepatocytes and activated KCs enhances fibrogenesis in HSCs. ROS are also involved in the activation of HSCs. In general, the OS triggered by ROS is critical for the development of NAFLD. Therefore, antioxidant therapy may be highly effective for the treatment of NAFLD.
In this review, we summarized reports on the application of classic antioxidants such as flavonoids, curcumin, vitamin E, metformin, and coenzyme Q10 in NALFD models. Although they target different signaling pathways, all these antioxidants ameliorate the pathological changes in NAFLD by reducing ROS. However, the most effective treatment for NAFLD may be to improve the quality of food consumed.
Finally, we also reviewed the role of OS in the development of NAFLD and progression to NASH and hepatic fibrosis and the application of antioxidants to NAFLD.
Funding
This research was funded by National Research Foundation of Korea [grant number: 2017R1A5A2015541 and 2019R1A2C1090178]; Ministry of Health and Welfare [grant number: HI20C0368]; Korean Food and Drug Administration [grant number: 20182MFDS425]; and by Regional Innovation Strategy grant from the Ministry of Education, South Korea [grant number: 2021RIS-001].
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Marcuccilli, M.; Chonchol, M. NAFLD and chronic kidney disease. Int. J. Mol. Sci. 2016, 17, 562. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.K. Similarities and differences between non-alcoholic fatty liver disease (NAFLD) & alcohol-associated liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6. [Google Scholar] [CrossRef]
- Toshikuni, N.; Tsutsumi, M.; Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393–8406. [Google Scholar] [CrossRef]
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative stress in NAFLD: Role of nutrients and food contaminants. Biomolelucles 2020, 10, 1702. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Massey, S.; Story, D.; Li, L. Metformin: An old drug with new applications. Int. J. Mol. Sci. 2018, 19, 2863. [Google Scholar] [CrossRef] [Green Version]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic fatty liver disease: Basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef] [PubMed]
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [Green Version]
- Simões, I.C.M.; Amorim, R.; Teixeira, J.; Karkucinska-Wieckowska, A.; Carvalho, A.; Pereira, S.P.; Simões, R.F.; Szymanska, S.; Dąbrowski, M.; Janikiewicz, J.; et al. The alterations of mitochondrial function during NAFLD progression—An independent effect of mitochondrial ROS production. Int. J. Mol. Sci. 2021, 22, 6848. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative stress in cancer cell metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Zarkovic, N. Roles and functions of ROS and RNS in cellular physiology and pathology. Cells 2020, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Martínez, M.C.; Andriantsitohaina, R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef]
- Rezzani, R.; Franco, C. Liver, oxidative stress and metabolic syndromes. Nutritients 2021, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Asp. Med. 2018, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.-R.; Chae, H.-J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Delaunay-Moisan, A.; Appenzeller-Herzog, C. The antioxidant machinery of the endoplasmic reticulum: Protection and signaling. Free. Radic. Biol. Med. 2015, 83, 341–351. [Google Scholar] [CrossRef]
- Araki, K.; Iemura, S.-I.; Kamiya, Y.; Ron, D.; Kato, K.; Natsume, T.; Nagata, K. Ero1-α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 2013, 202, 861–874. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wu, X.; Wang, P.; Zhang, S.; Sun, L.; Zhao, Y.; Zeng, G.; Liu, B.; Xu, G.; Liu, H.; et al. Homocysteine promotes hepatic steatosis by activating the adipocyte lipolysis in a HIF1α-ERO1α-dependent oxidative stress manner. Redox Biol. 2020, 37, 101742. [Google Scholar] [CrossRef]
- Al-Saleh, F.; Khashab, F.; Fadel, F.; Al-Kandari, N.; Al-Maghrebi, M. Inhibition of NADPH oxidase alleviates germ cell apoptosis and ER stress during testicular ischemia reperfusion injury. Saudi J. Biol. Sci. 2020, 27, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, R.; Kato, M.; Miyagawa, S.; Kamata, T. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2013, 33, 4431–4438. [Google Scholar] [PubMed]
- Gabbia, D.; Cannella, L.; De Martin, S. The role of oxidative stress in NAFLD–NASH–HCC transition—Focus on NADPH oxidases. Biomedicines 2021, 9, 687. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef]
- Chen, C.; Li, L.; Zhou, H.J.; Min, W. The role of NOX4 and TRX2 in angiogenesis and their potential cross-talk. Antioxidants 2017, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arter. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Qiu, T.; Yao, X.; Jiang, L.; Wang, N.; Jiang, J.; Jia, X.; Wei, S.; Zhang, J.; Zhu, Y.; et al. IRE1α/NOX4 signaling pathway mediates ROS-dependent activation of hepatic stellate cells in NaAsO2-induced liver fibrosis. J. Cell. Physiol. 2021, 236, 1469–1480. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Kehrer, I.; Kozlov, A.V.; Haller, M.; Redl, H.; Hermann, M.; Grimm, M.; Troppmair, J. Mitochondrial ROS production under cellular stress: Comparison of different detection methods. Anal. Bioanal. Chem. 2011, 400, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.J.; DeVylder, J.E.; Fedina, L. Police violence among adults diagnosed with mental disorders. Health Soc. Work 2020, 45, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Völkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. BioFactors 2009, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.N.; Walker, C.L. The peroxisome as a cell signaling organelle. Curr. Opin. Cell Biol. 2016, 39, 109–112. [Google Scholar] [CrossRef] [Green Version]
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA derived from hepatic peroxisomal β-oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol. Cell 2020, 79, 30–42.e4. [Google Scholar] [CrossRef]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef] [Green Version]
- Reddy, J.K. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1333–G1339. [Google Scholar] [CrossRef]
- Nagano, T.; Yamao, S.; Terachi, A.; Yarimizu, H.; Itoh, H.; Katasho, R.; Kawai, K.; Nakashima, A.; Iwasaki, T.; Kikkawa, U.; et al. d-amino acid oxidase promotes cellular senescence via the production of reactive oxygen species. Life Sci. Alliance 2019, 2, e201800045. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; Cappelletti, P.; Murtas, G. Biochemical properties of human D-amino acid oxidase variants and their potential significance in pathologies. Front. Mol. Biosci. 2018, 5, 55. [Google Scholar] [CrossRef] [Green Version]
- Koga, R.; Miyoshi, Y.; Sakaue, H.; Hamase, K.; Konno, R. Mouse d-amino-acid oxidase: Distribution and physiological substrates. Front. Mol. Biosci. 2017, 4, 82. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Okamura, T.; Kasai, N.; Hori, Y.; Summer, K.H.; Konno, R. D-amino-acid oxidase is involved in d-serine-induced nephrotoxicity. Chem. Res. Toxicol. 2005, 18, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Xu, Y.; Zhang, C.; Wang, N.; Li, H.; Feng, Y. Green tea and epigallocatechin gallate (EGCG) for the management of nonalcoholic fatty liver diseases (NAFLD): Insights into the role of oxidative stress and antioxidant mechanism. Antioxidants 2021, 10, 1076. [Google Scholar] [CrossRef]
- Mazzolini, G.; Sowa, J.P.; Atorrasagasti, C.; Kücükoglu, Ö.; Syn, W.K.; Canbay, A. Significance of simple steatosis: An update on the clinical and molecular evidence. Cells 2020, 9, 2458. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Giorgio, V.; Prono, F.; Graziano, F.; Nobili, V. Pediatric non alcoholic fatty liver disease: Old and new concepts on development, progression, metabolic insight and potential treatment targets. BMC Pediatr. 2013, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abinaya, M.; Prabhakaran, S.; Byoung, R.J. Role of reactive oxygen species signaling in cell proliferation and differentiation. React. Oxyg. Species Plants 2017, 319–329. [Google Scholar]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial reactive oxygen species and mitophagy: A complex and nuanced relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Middleton, P.; Vergis, N. Mitochondrial dysfunction and liver disease: Role, relevance, and potential for therapeutic modulation. Ther. Adv. Gastroenterol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk sac-derived erythro-myeloid progenitors. Exp. Hematol. 2015, 43, S64. [Google Scholar] [CrossRef]
- Bailey, S.M.; Cunningham, C.C. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability in fresh, isolated rat hepatocytes. Hepatology 1998, 28, 1318–1326. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, Y.; Li, Q.; Xu, M.; Bai, J.; Wu, S. Resveratrol improves alcoholic fatty liver disease by downregulating HIF-1α expression and mitochondrial ROS production. PLoS ONE 2017, 12, e0183426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer cells in non-alcoholic fatty liver disease: Friend or foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, R.J.; Rüegg, J.C.; Morano, I. Counting target molecules by exponential polymerase chain reaction: Copy number of mitochondrial DNA in rat tissues. Biochem. Biophys. Res. Commun. 1992, 183, 553–559. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.T.; Nair, L.A.; Livingston, K.; Fu, X.R.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Qin, S.; Yin, J.; Huang, K. Free fatty acids increase intracellular lipid accumulation and oxidative stress by modulating PPARα and SREBP-1c in L-02 cells. Lipids 2016, 51, 797–805. [Google Scholar] [CrossRef]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendl, N.; Occhipinti, A.; Muller, M.; Wild, P.; Dikic, I.; Reichert, A.S. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J. Cell Sci. 2011, 124, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Von Stockum, S.; Nardin, A.; Schrepfer, E.; Ziviani, E. Mitochondrial dynamics and mitophagy in Parkinson’s disease: A fly point of view. Neurobiol. Dis. 2016, 90, 58–67. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, parkin, and mitochondrial quality control: What can we learn about Parkinson’s disease pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.M.J.; Moreira, P.I.; Ambrosio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Autophagy and mitophagy in cellular damage control. Redox Biol. 2013, 1, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Shirihai, O.S. The Interplay between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.; Zong, Y.; Yang, X. DHA protects hepatocytes from oxidative injury through GPR120/ERK-mediated mitophagy. Int. J. Mol. Sci. 2021, 22, 5675. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and mechanisms of mitophagy in liver diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Yilancioglu, K.; Cokol, M.; Pastirmaci, I.; Erman, B.; Cetiner, S. Oxidative Stress Is a Mediator for Increased Lipid Accumulation in a Newly Isolated Dunaliella salina Strain. PLoS ONE 2014, 9, e91957. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.L.; Hu, Y.Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.F.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- She, C.; Zhu, L.Q.; Zhen, Y.F.; Wang, X.D.; Dong, Q.R. Activation of AMPK protects against hydrogen peroxide-induced osteoblast apoptosis through autophagy induction and NADPH maintenance: New implications for osteonecrosis treatment? Cell. Signal. 2014, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Huang, W.; Gu, J.; Du, X.; Lei, L.; Yuan, X.; Sun, G.; Wang, Z.; Li, X.; Liu, G. SREBP-1c overactivates ROS-mediated hepatic NF-kappaB inflammatory pathway in dairy cows with fatty liver. Cell. Signal. 2015, 27, 2099–2109. [Google Scholar] [CrossRef]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, T.; Farrar, S.; Ji, L.B.; Liu, T.Y.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Mello, T.; Zanieri, F.; Ceni, E.; Galli, A. Oxidative stress in the healthy and wounded hepatocyte: A cellular organelles perspective. Oxidative Med. Cell. Longev. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Homma, T.; Kurahashi, T.; Kang, E.S.; Fujii, J. Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem. Biophys. Res. Commun. 2015, 464, 229–235. [Google Scholar] [CrossRef]
- Li, S.; Fu, L.; Tian, T.; Deng, L.; Li, H.; Xia, W.; Gong, Q. Disrupting SOD1 activity inhibits cell growth and enhances lipid accumulation in nasopharyngeal carcinoma. Cell Commun. Signal. 2018, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The dual role of Nrf2 in nonalcoholic fatty liver disease: Regulation of antioxidant defenses and hepatic lipid metabolism. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Han, J.H.; Park, M.H.; Myung, C.S. Garcinia cambogia ameliorates non-alcoholic fatty liver disease by inhibiting oxidative stress-mediated steatosis and apoptosis through NRF2-ARE activation. Antioxidants 2021, 10, 1226. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef]
- Poelstra, K.; Schuppan, D. Targeted therapy of liver fibrosis/cirrhosis and its complications. J. Hepatol. 2011, 55, 726–728. [Google Scholar] [CrossRef] [Green Version]
- Czaja, M.J.; Liu, H.; Wang, Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology 2003, 37, 1405–1413. [Google Scholar] [CrossRef]
- Nomoto, K.; Tsuneyama, K.; Takahashi, H.; Murai, Y.; Takano, Y. Cytoplasmic Fine Granular Expression of 8-hydroxydeoxyguanosine Reflects Early Mitochondrial Oxidative DNA Damage in Nonalcoholic Fatty Liver Disease. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B.; Mansouri, A. Mitochondrial injury in steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2004, 16, 1095–1105. [Google Scholar] [CrossRef]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Devkar, R.V.; Nammi, S. Oxidative Stress in Liver Diseases: Pathogenesis, Prevention, and Therapeutics. Oxidative Med. Cell. Longev. 2017, 2017, 1–2. [Google Scholar] [CrossRef]
- Bouwens, L.; Baekeland, M.; de Zanger, R.; Wisse, E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 1986, 6, 718–722. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.X.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L.C. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.N.; Simoes, I.; Rosa, H.; Khan, S.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; Khan, R.; Wieckowska, K. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells 2019, 8, 1222. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.L.; Hoque, R.; Ouyang, X.S.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.S.; Liu, Y.; An, W.S.; Song, J.W.; Zhang, Y.F.; Zhao, X.X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.P.; Wu, J.X.; Zhang, X.W.; Sun, L.J.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.J.; Wu, J.; Zhang, X.F.; Li, X.H.; Wu, X.W.; Zhao, Y.; Ren, J.A. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis. 2021, 12, 673. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019, 29, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.J.; Ou, Z.B.; Cai, C.; Li, P.Z.; Gong, J.P.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.-L.; Xiao, M.; Yang, R.; Xie, K.-Q. Critical roles of Kupffer cells in the pathogenesis of alcoholic liver disease: From basic science to clinical trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef] [PubMed]
- Luckey, S.W.; Petersen, D.R. Metabolism of 4-hydroxynonenal by rat Kupffer cells. Arch. Biochem. Biophys. 2001, 389, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, P.; Derdak, Z.; Sheets, A.; Sabo, E.; Berthiaume, E.P.; Resnick, M.B.; Wands, J.R.; Paragh, G.; Baffy, G. Lack of UCP2 reduces fas-mediated liver injury in ob/ob mice and reveals importance of cell-specific UCP2 expression. Hepatolgy 2006, 44, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Evans, Z.P.; Ellett, J.D.; Schmidt, M.G.; Schnellmann, R.G.; Chavin, K.D. Mitochondrial uncoupling protein-2 mediates steatotic liver injury following ischemia/reperfusion. J. Biol. Chem. 2008, 283, 8573–8579. [Google Scholar] [CrossRef] [Green Version]
- Pierelli, G.; Stanzione, R.; Forte, M.; Migliarino, S.; Perelli, M.; Volpe, M.; Rubattu, S. Uncoupling protein 2: A key player and a potential therapeutic target in vascular diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH: Ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Onuma, H.; Bai, X.; Medvedev, A.V.; Misukonis, M.; Weinberg, J.B.; Cao, W.H.; Robidoux, J.; Floering, L.M.; Daniel, K.W.; et al. Persistent nuclear factor-kappa B activation in UCP2-/- mice leads to enhanced nitric oxide and inflammatory cytokine production. J. Biol. Chem. 2005, 280, 19062–19069. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling—Respiratory burst in macrophage signaling. Am. J. Resp. Crit. Care 2002, 166, S4–S8. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Zhang, W.F.; Wu, Y.K.; Mu, D.; Gong, J.P.; Wu, C.X.; Huang, C. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar]
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-kappaB signaling pathway. J. Gastroenterol. Hepatol. 2020, 35, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Nolan, C.J.; Larter, C.Z. Lipotoxicity: Why do saturated fatty acids cause and monounsaturates protect against it? J. Gastroenterol. Hepatol. 2009, 24, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.L.; Vorac, J.; Hofmann, K.; Meiser, P.; Unterweger, I.; Kuerschner, L.; Weighardt, H.; Förster, I.; Thiele, C. Kupffer cells sense free fatty acids and regulate hepatic lipid metabolism in high-fat diet and inflammation. Cells 2020, 9, 2258. [Google Scholar] [CrossRef]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative stress in cardiovascular diseases: Involvement of Nrf2 antioxidant redox signaling in macrophage foam cells formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef] [Green Version]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.-L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Scott, C.L.; Guilliams, M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018, 69, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Yeligar, S.M.; Machida, K.; Kalra, V.K. Ethanol-induced HO-1 and NQO1 Are Differentially Regulated by HIF-1α and Nrf2 to Attenuate Inflammatory Cytokine Expression. J. Biol. Chem. 2010, 285, 35359–35373. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzimato, V.; Jager, J.; Chen, P.; Morgantini, C.; Levi, L.; Barreby, E.; Sulen, A.; Oses, C.; Willerbrords, J.; Xu, C.; et al. Liver macrophages inhibit the endogenous antioxidant response in obesity-associated insulin resistance. Sci. Transl. Med. 2020, 12, e9709. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ni, M.; Tian, Y.; Wang, H.; Qiu, J.; You, W.; Wei, S.; Shi, Y.; Zhou, J.; Cheng, F.; et al. Myeloid Nrf2 deficiency aggravates non-alcoholic steatohepatitis progression by regulating YAP-mediated NLRP3 inflammasome signaling. iScience 2021, 24, 102427. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Warabi, E.; Okada, K.; Yanagawa, T.; Ishii, T.; Kose, K.; Tokushige, K.; Ishige, K.; Mizokami, Y.; Yamagata, K.; et al. Deletion of both p62 and Nrf2 spontaneously results in the development of nonalcoholic steatohepatitis. Exp. Anim. 2018, 67, 201–218. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Priyadarshi, R.N.; Anand, U. Non-alcoholic fatty liver disease: Growing burden, adverse outcomes and associations. J. Clin. Transl. Hepatol. 2019, 8, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Syn, W.K. Role of metabolism in hepatic stellate cell activation and fibrogenesis. Front. Cell Dev. Biol. 2018, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Seo, W.; Eun, H.S.; Kim, S.Y.; Yi, H.S.; Lee, Y.S.; Park, S.H.; Jang, M.J.; Jo, E.; Kim, S.C.; Han, Y.M.; et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 2016, 64, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e8. [Google Scholar] [CrossRef]
- Ou, Q.; Weng, Y.; Wang, S.; Zhao, Y.; Zhang, F.; Zhou, J.; Wu, X. Silybin alleviates hepatic steatosis and fibrosis in NASH mice by inhibiting oxidative stress and involvement with the Nf-κB pathway. Dig. Dis. Sci. 2018, 63, 3398–3408. [Google Scholar] [CrossRef]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- De Bleser, P.J.; Xu, G.; Rombouts, K.; Rogiers, V.; Geerts, A. Glutathione levels discriminate between oxidative stress and transforming growth factor-beta signaling in activated rat hepatic stellate cells. J. Biol. Chem. 1999, 274, 33881–338817. [Google Scholar] [CrossRef] [Green Version]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH Oxidases in Liver Fibrosis. Antioxid. Redox Signal. 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, I.C.; Eun, J.R. Pharmacologic therapy for nonalcoholic steatohepatitis focusing on pathophysiology. Yeungnam Univ. J. Med. 2019, 36, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schröder, K.; Brandes, R.; Devaraj, S.; Török, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, K.; Gorin, Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 2012, 12, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodríguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef]
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Osterreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010, 139, 1375–1384.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Sohail, M.A.; Gomes, D.A.; Hashmi, A.; Nagata, J.; Sutterwala, F.S.; Mahmood, S.; Jhandier, M.N.; Shi, Y.; Flavell, R.A.; et al. Inflammasome-mediated regulation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1248–G1257. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.M.; Yang, R.Q.; Li, Y.; Ning, Z.W.; Zhang, L.L.; Zhou, G.S.; Luo, W.; Li, D.H.; Chen, M.Y.; Pan, M.X.; et al. Angiotensin-(1–7) improves liver fibrosis by regulating the NLRP3 inflammasome via redox balance modulation. Antioxid. Redox Signal. 2016, 24, 795–812. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Feng, D.; Zhao, H.; Lin, M.; Hu, Y.; Zhang, N.; Lv, L.; Gao, Z.; Zhai, X.; et al. p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics 2019, 9, 1510–1522. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestigiacomo, V.; Suter-Dick, L. Nrf2 protects stellate cells from Smad-dependent cell activation. PLoS ONE 2018, 13, e0201044. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms that Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Kaji, K.; Sato, S.; Ogawa, H.; Takagi, H.; Takaya, H.; Kawaratani, H.; Moriya, K.; Namisaki, T.; Akahane, T.; et al. Sulforaphane ameliorates ethanol plus carbon tetrachloride-induced liver fibrosis in mice through the Nrf2-mediated antioxidant response and acetaldehyde metabolization with inhibition of the LPS/TLR4 signaling pathway. J. Nutr. Biochem. 2021, 89, 108573. [Google Scholar] [CrossRef]
- Kim, M.; Yang, S.-G.; Kim, J.M.; Lee, J.-W.; Kim, Y.S.; Lee, J.I. Silymarin suppresses hepatic stellate cell activation in a dietary rat model of non-alcoholic steatohepatitis: Analysis of isolated hepatic stellate cells. Int. J. Mol. Med. 2012, 30, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.G.I.; Jain, P.; Khan, Z.K. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peluso, I.; Miglio, C.; Morabito, G.; Ioannone, F.; Serafini, M. Flavonoids and immune function in human: A systematic review. Crit. Rev. Food Sci. Nutr. 2015, 55, 383–395. [Google Scholar] [CrossRef]
- Wang, K.; Tan, W.; Liu, X.; Deng, L.; Huang, L.; Wang, X.; Gao, X. New insight and potential therapy for NAFLD: CYP2E1 and flavonoids. Biomed. Pharmacother. 2021, 137, 111326. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.; Zhu, Q.Y.; Tsang, D.; Huang, Y. Degradation of green tea catechins in tea drinks. J. Agric. Food Chem. 2000, 49, 477–482. [Google Scholar] [CrossRef]
- Kuzu, N.; Bahcecioglu, I.H.; Dagli, A.F.; Ozercan, I.H.; Ustündag, B.; Sahin, K. Epigallocatechin gallate attenuates experimental non-alcoholic steatohepatitis induced by high fat diet. J. Gastroenterol. Hepatol. 2008, 23, e465–e470. [Google Scholar] [CrossRef]
- Xiao, J.; Ho, C.T.; Liong, E.C.; Nanji, A.A.; Leung, T.M.; Lau, T.Y.H.; Fung, M.L.; Tipoe, G.L. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 2014, 53, 187–199. [Google Scholar] [CrossRef]
- Niu, Y.; Na, L.; Feng, R.; Gong, L.; Zhao, Y.; Li, Q.; Li, Y.; Sun, C. The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age-associated inflammation and oxidative stress in healthy rats. Aging Cell 2013, 12, 1041–1049. [Google Scholar] [CrossRef]
- Shishodia, S. Molecular mechanisms of curcumin action: Gene expression. BioFactors 2012, 39, 37–55. [Google Scholar] [CrossRef]
- Pan, M.H.; Lai, C.S.; Tsai, M.L.; Ho, C.T. Chemoprevention of nonalcoholic fatty liver disease by dietary natural compounds. Mol. Nutr. Food Res. 2013, 58, 147–171. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, Y.; Zhang, X.; Aa, J.; Wang, G.; Xie, Y. Curcumin regulates endogenous and exogenous metabolism via Nrf2-FXR-LXR pathway in NAFLD mice. Biomed. Pharmacother. 2018, 105, 274–281. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; De Matteo, E.; Baz, P.; Lucero, D.; Garcia, C.C.; Gonzales Ballerga, E.; Daruich, J.; Sorda, J.A.; Wald, M.R.; Cherñavsky, A.C. New evidence for the therapeutic potential of curcumin to treat nonalcoholic fatty liver disease in humans. PLoS ONE 2017, 12, e0172900. [Google Scholar] [CrossRef]
- Lee, D.E.; Lee, S.J.; Kim, S.J.; Lee, H.S.; Kwon, O.S. Curcumin ameliorates nonalcoholic fatty liver disease through inhibition of O-GlcNAcylation. Nutrients 2019, 11, 2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Argo, C.K.; Al-Osaimi, A.M.S.; Caldwell, S.H. Therapy of NAFLD: Antioxidants and cytoprotective agents. J. Clin. Gastroenterology 2006, 40 (Suppl. S1), S51–S60. [Google Scholar]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The role of vitamin E in the treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.Y.; Yeung, S.F.; Park, H.J.; Volek, J.S.; Bruno, R.S. Dietary alpha- and gamma-tocopherol supplementation attenuates lipopolysaccharide-induced oxidative stress and inflammatory-related responses in an obese mouse model of nonalcoholic steatohepatitis. J. Nutr. Biochem. 2010, 21, 1200–1206. [Google Scholar] [CrossRef]
- Iida, C.; Fujii, K.; Koga, E.; Washino, Y.; Kitamura, Y.; Ichi, I.; Abe, K.; Matsura, T.; Kojo, S. Effect of α-tocopherol on carbon tetrachloride intoxication in the rat liver. Arch. Toxicol. 2009, 83, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.M.; Wu, W.J.; Fu, N.; Liang, B.L.; Wang, R.Q.; Li, L.X.; Zhao, S.X.; Zhao, J.M.; Yu, J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 1121–1131. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Geng, Y.; Hernandez Villanueva, A.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165621. [Google Scholar] [CrossRef]
- Geerling, J.J.; Boon, M.R.; van der Zon, G.C.; van den Berg, S.A.; van den Hoek, A.M.; Lombès, M.; Princen, H.M.; Havekes, L.M.; Rensen, P.C.; Guigas, B. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes 2014, 63, 880–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karise, I.; Ornellas, F.; Barbosa-da-Silva, S.; Matsuura, C.; Del Sol, M.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Liver and Metformin: Lessons of a fructose diet in mice. Biochim. Open 2017, 4, 19–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Xiao, H. Metformin actions on the liver: Protection mechanisms emerging in hepatocytes and immune cells against NASH-related HCC. Int. J. Mol. Sci. 2021, 22, 5016. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Mariscal, F.M.; Arenas-de Larriva, A.P.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 supplementation for the reduction of oxidative stress: Clinical implications in the treatment of chronic diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, X.; Xue, H.; Zhang, P.; Fang, W.; Chen, X.; Ling, W. Coenzyme Q10 attenuates high-fat diet-induced non-alcoholic fatty liver disease through activation of the AMPK pathway. Food Funct. 2019, 10, 814–823. [Google Scholar] [CrossRef]
- Perumpail, B.J.; Li, A.A.; Iqbal, U.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. Potential therapeutic benefits of herbs and supplements in patients with NAFLD. Diseases 2018, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botham, K.M.; Napolitano, M.; Bravo, E. The emerging role of disturbed CoQ metabolism in nonalcoholic fatty liver disease development and progression. Nutritients 2015, 7, 9834–9846. [Google Scholar] [CrossRef] [Green Version]
- Lavine, J.E. Vitamin E treatment of nonalcoholic steatohepatitis in children: A pilot study. J. Pediatr. 2000, 136, 734–738. [Google Scholar] [CrossRef]
- El Hadi, H.; Vettor, R.; Rossato, M. Vitamin E as a treatment for nonalcoholic fatty liver disease: Reality or myth? Antioxidants 2018, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Yoneda, M.; Nakamura, K.; Makino, I.; Terano, A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Aliment. Pharmacol. Ther. 2001, 15, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [Green Version]
- Vilà, L.; Rebollo, A.; Ađalsteisson, G.S.; Alegret, M.; Merlos, M.; Roglans, N.; Laguna, J.C. Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment. Toxicol. Appl. Pharmacol. 2011, 251, 32–40. [Google Scholar] [CrossRef]
- Foster, T.; Budoff, M.J.; Saab, S.; Ahmadi, N.; Gordon, C.; Guerci, A.D. Atorvastatin and Antioxidants for the Treatment of Nonalcoholic Fatty Liver Disease: The St Francis Heart Study Randomized Clinical Trial. Am. J. Gastroenterol. 2011, 106, 71–77. [Google Scholar] [CrossRef]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism 2008, 57, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Sohet, F.M.; Neyrinck, A.M.; Pachikian, B.D.; de Backer, F.C.; Bindels, L.B.; Niklowitz, P.; Menke, T.; Cani, P.D.; Delzenne, N.M. Coenzyme Q10 supplementation lowers hepatic oxidative stress and inflammation associated with diet-induced obesity in mice. Biochem. Pharmacol. 2009, 78, 1391–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheibi, S.; Ghaleh, H.E.G.; Motlagh, B.M.; Azarbayjani, A.F.; Zarei, L. Therapeutic effects of curcumin and ursodexycholic acid on non-alcoholic fatty liver disease. Biomed. Pharmacother. 2019, 115, 108938. [Google Scholar] [CrossRef]
- Li, L.; Hai, J.; Li, Z.; Zhang, Y.; Peng, H.; Li, K.; Weng, X. Resveratrol modulates autophagy and NF-κB activity in a murine model for treating non-alcoholic fatty liver disease. Food Chem. Toxicol. 2014, 63, 166–173. [Google Scholar] [CrossRef]
- Elgebaly, A.; Radwan, I.A.; AboElnas, M.M.; Ibrahim, H.H.; Eltoomy, M.F.; Atta, A.A.; Mesalam, H.A.; Sayed, A.A.; Othman, A.A. Resveratrol Supplementation in Patients with Non-Alcoholic Fatty Liver Disease: Systematic Review and Meta-analysis. J. Gastrointest. Liver Dis. 2017, 26, 59–67. [Google Scholar] [CrossRef]
- Bujanda, L.; Hijona, E.; Larzabal, M.; Beraza, M.; Aldazabal, P.; García-Urkia, N.; Sarasqueta, C.; Cosme, A.; Irastorza, B.; González, A.; et al. Resveratrol inhibits nonalcoholic fatty liver disease in rats. BMC Gastroenterol. 2008, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Liu, H.; Li, C. Dietary regulation of oxidative stress in chronic metabolic diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef]
- Parks, E.; Yki-Jarvinen, H.; Hawkins, M. Out of the frying pan: Dietary saturated fat influences nonalcoholic fatty liver disease. J. Clin. Investig. 2017, 127, 454–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, S.C.; Bernal, W.; Dasarathy, S.; Merli, M.; Plank, L.D.; Schütz, T.; Plauth, M. ESPEN practical guideline: Clinical nutrition in liver disease. Clin. Nutr. 2020, 39, 3533–3562. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Corey, K.E.; Lim, J.K. AGA clinical practice update on lifestyle modification using diet and exercise to achieve weight loss in the management of nonalcoholic fatty liver disease: Expert review. Gastroenterology 2021, 160, 912–918. [Google Scholar] [CrossRef]
- Houttu, V.; Csader, S.; Nieuwdorp, M.; Holleboom, A.G.; Schwab, U. Dietary interventions in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Front. Nutr. 2021, 8, 8. [Google Scholar] [CrossRef]
- Berná, G.; Romero-Gomez, M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int. 2020, 40 (Suppl. S1), 102–108. [Google Scholar] [CrossRef] [Green Version]
- Suárez, M.; Boqué, N.; Del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean diet and multi-ingredient-based interventions for the management of non-alcoholic fatty liver disease. Nutritients 2017, 9, 1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Major source of ROS production in Hepatic cell. The catalytic process of oxidoreductase Ero1 and NADPH oxidase (NOX) in the endoplasmic reticulum generates ROS. During cellular respiration, mitochondria transfer electrons to oxygen and generate ROS as a byproduct of oxidative oxidation. Mitochondrial ROS are mostly produced during oxidative phosphorylation of electron transport chains (ETCs) present in the inner mitochondrial membrane. The main cause of ROS generation in peroxisomes is peroxisomal β-oxidation. Acyl-CoA is converted to Enoyl-CoA by acyl-CoA oxidase containing FAD, which provides electrons directly to oxygen to produce H₂O₂.
Figure 1.
Major source of ROS production in Hepatic cell. The catalytic process of oxidoreductase Ero1 and NADPH oxidase (NOX) in the endoplasmic reticulum generates ROS. During cellular respiration, mitochondria transfer electrons to oxygen and generate ROS as a byproduct of oxidative oxidation. Mitochondrial ROS are mostly produced during oxidative phosphorylation of electron transport chains (ETCs) present in the inner mitochondrial membrane. The main cause of ROS generation in peroxisomes is peroxisomal β-oxidation. Acyl-CoA is converted to Enoyl-CoA by acyl-CoA oxidase containing FAD, which provides electrons directly to oxygen to produce H₂O₂.

Figure 2.
The mechanism of ROS-mediated oxidative stress and lipid metabolism in hepatocytes. In the NAFLD patients, the overload intake of free fatty acids increases fatty acid β-oxidation and electron transport chain activity in the mitochondria. This ultimately leads to an increased release of ROS as byproduct of metabolism. The lots of ROS can directly target mitochondria, resulting in decreased energy metabolism, increased release of mtDNA and mitochondrial dysfunction. At the same time, mtROS leads to disturbance of mitochondrial membrane potential. High mitochondrial membrane potential is not friendly to the occurrence of mitophagy. The reduction of mitophagy reduces the clearance of damaged mitochondria and indirectly increases the fat synthesis. Moreover, high levels of ROS activate AMPK, SREBP1, NF-kB, JNK/cJun, and downregulate NRF2. These protein kinases and transcription factors regulate lipid metabolism, inflammation, antioxidant capacity and hepatocytes apoptosis. Finally, with the increase in insulin resistance, the synthesis of fat is intensified in hepatocytes.
Figure 2.
The mechanism of ROS-mediated oxidative stress and lipid metabolism in hepatocytes. In the NAFLD patients, the overload intake of free fatty acids increases fatty acid β-oxidation and electron transport chain activity in the mitochondria. This ultimately leads to an increased release of ROS as byproduct of metabolism. The lots of ROS can directly target mitochondria, resulting in decreased energy metabolism, increased release of mtDNA and mitochondrial dysfunction. At the same time, mtROS leads to disturbance of mitochondrial membrane potential. High mitochondrial membrane potential is not friendly to the occurrence of mitophagy. The reduction of mitophagy reduces the clearance of damaged mitochondria and indirectly increases the fat synthesis. Moreover, high levels of ROS activate AMPK, SREBP1, NF-kB, JNK/cJun, and downregulate NRF2. These protein kinases and transcription factors regulate lipid metabolism, inflammation, antioxidant capacity and hepatocytes apoptosis. Finally, with the increase in insulin resistance, the synthesis of fat is intensified in hepatocytes.

Figure 3.
The mechanism of ROS involved in pro-inflammatory Kupffer cells. Fatty acid intake increases lipid accumulation in hepatocytes, whereas oxidative stress involves the release of mtDNA from damaged hepatocytes. KCs activated by mtDNA promotes the secretion of pro-inflammatory factors through the cGAS/STING axis and is dependent on the NF-kB pathway. LPS-mediated inflammation through activation of MAPK and NF-kB pathways. Meanwhile, LPS activates TLR4 to promote NOX2 production of ROS. Large amounts of ROS increase MAPK and NF-kB transcription and promote production of inflammatory cytokines as well as (IL-1β, TNF-α, and IL-6, etc.). Mitochondria are the main organelle for ROS production, and UCP2 localized in the inner mitochondrial membrane increase proton transfer to downregulate mitochondrial membrane potential to control ROS levels. NRF2 regulate negatively ROS by increasing the production of antioxidant factors.
Figure 3.
The mechanism of ROS involved in pro-inflammatory Kupffer cells. Fatty acid intake increases lipid accumulation in hepatocytes, whereas oxidative stress involves the release of mtDNA from damaged hepatocytes. KCs activated by mtDNA promotes the secretion of pro-inflammatory factors through the cGAS/STING axis and is dependent on the NF-kB pathway. LPS-mediated inflammation through activation of MAPK and NF-kB pathways. Meanwhile, LPS activates TLR4 to promote NOX2 production of ROS. Large amounts of ROS increase MAPK and NF-kB transcription and promote production of inflammatory cytokines as well as (IL-1β, TNF-α, and IL-6, etc.). Mitochondria are the main organelle for ROS production, and UCP2 localized in the inner mitochondrial membrane increase proton transfer to downregulate mitochondrial membrane potential to control ROS levels. NRF2 regulate negatively ROS by increasing the production of antioxidant factors.

Figure 4.
The mechanism of ROS in activation of stellate cells. TGF-β is released from activated KCs, the major cell type for cytokine production during NASH. the TGF-β-SMAD2/3 axis is activated in HSCs, which upregulates NOX4 expression to increase ROS production. The additional ROS directly drives the production of extracellular matrix (collagen-I and α-SMA) in HSCs. On the other side, TGF-β elevate NLRP3 activation to leads ROS production. The concomitant release of ROS from mitochondria exacerbates the activation of HSCs. Although moderate amounts of ROS can activate the dissociation of NRF2 and KEAP1 and drive NRF2 transcription, the NRF2-regulated antioxidant system can hardly resist the impact of large amounts of ROS in cells.
Figure 4.
The mechanism of ROS in activation of stellate cells. TGF-β is released from activated KCs, the major cell type for cytokine production during NASH. the TGF-β-SMAD2/3 axis is activated in HSCs, which upregulates NOX4 expression to increase ROS production. The additional ROS directly drives the production of extracellular matrix (collagen-I and α-SMA) in HSCs. On the other side, TGF-β elevate NLRP3 activation to leads ROS production. The concomitant release of ROS from mitochondria exacerbates the activation of HSCs. Although moderate amounts of ROS can activate the dissociation of NRF2 and KEAP1 and drive NRF2 transcription, the NRF2-regulated antioxidant system can hardly resist the impact of large amounts of ROS in cells.


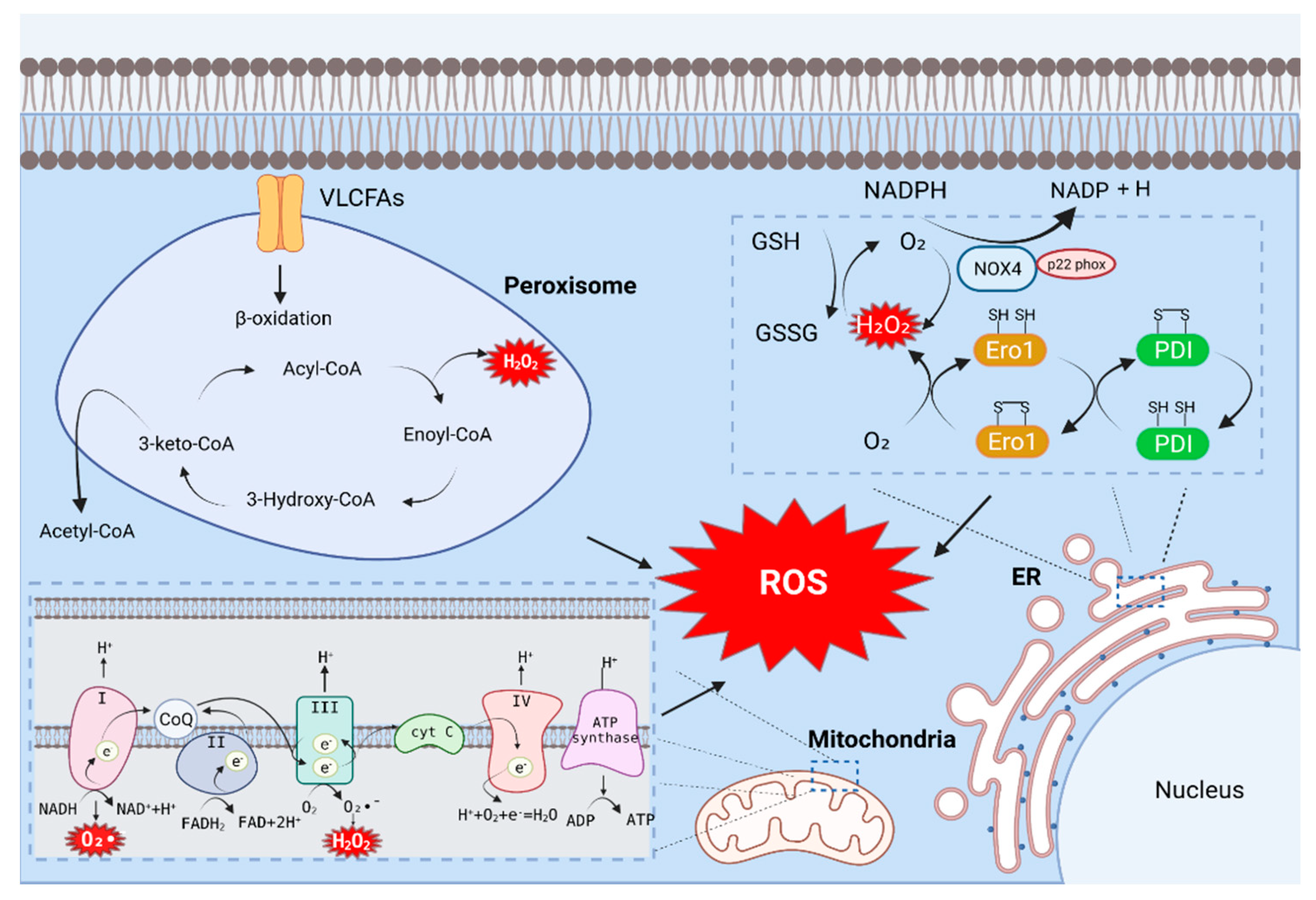{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Therapeutic compounds based on antioxidant properties in NAFLD.
| Name | Mechanism | Effects | CLD | Stage | Ref. | PubChem CID |
|---|---|---|---|---|---|---|
| α-tocopherol Atorvastatin | Inhibition of MAPK (JNK/p38 MAPK) signaling and NF-κB activation. | Free radical stabilization Replenish liver glutathione, reduce steatosis, improve inflammation, hepatic stellate cell activation, collagen mRNA expression and fibrosis. | NASH | Clinical phase 2 | [190] [191] [192] [193] | 14985 |
| Inhibition of NF-Κb signaling and HMG-CoA reductase. | Reduction activity of fatty acid β-oxidation, activation of ChREBP, plasma ALT levels, and inflammation in liquid fructose-fed mice. | NAFLD NASH | Clinical phase 2 | [194] [195] [196] | 60823 | |
| Epigallocatechin-3-gallate (EGCG) | Inhibition of oxidative stress Inhibition of NF-κB/AKT signailing Inhibition of TGF/ SMAD signaling | Increased SOD activity in serum and liver tissue. Reduction of liver fibrosis in NASH rodent model. Reduction TNF-a and IL-6 mRNA expression in serum and liver tissue. | NASH | Clinical phase 1 | [168] [169] [170] | 65064 |
| Metformin | AMPK and NRF-2 signaling activation | Inhibition of ROS production Increase in SOD2 activation Decrease lipid accumulation in liver and TG level in serum | NAFLD NASH | Clinical phase 4 | [182] [183] [184] | 4091 |
| Coenzyme Q | AMPK signaling activation | Inhibition of NADPH oxidase expression Inhibition of liver steatosis Reduction of TNF-a, IL-6 mRNA expression | NAFLD NASH | Clinical phase | [187] [188] [189] [197] | 5281915 |
| Curcumin | AMPK/ACC signaling activation | Induction of TAC, GSH-Px and SOD level Inhibition lipid accumulation in liver Inhibition hepatic inflammation and fibrosis Reduction of ER stress | NAFLD NASH | Clinical phase 4 | [174] [175] [198] | 969516 |
| Resveratrol | Activation of autophagy Inhibition of NF-Κb signaling | Induction of superoxide dismutase(SOD), glutathione peroxidase and catalase Regulation of lipid accumulation through autophagy Inhibition of hepatic inflammation | NAFLD, NASH | Clinical phase 2 | [199] [200] [201] | 445154 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ma, Y.; Lee, G.; Heo, S.-Y.; Roh, Y.-S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants 2022, 11, 91. https://doi.org/10.3390/antiox11010091
AMA Style
Ma Y, Lee G, Heo S-Y, Roh Y-S. Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease. Antioxidants. 2022; 11(1):91. https://doi.org/10.3390/antiox11010091
Chicago/Turabian StyleMa, Yuanqiang, Gyurim Lee, Su-Young Heo, and Yoon-Seok Roh. 2022. "Oxidative Stress Is a Key Modulator in the Development of Nonalcoholic Fatty Liver Disease" Antioxidants 11, no. 1: 91. https://doi.org/10.3390/antiox11010091
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.